
ERF: Il Gene Sospetto che Riscrive la Sindrome di Noonan (Senza Chiudere le Suture!)
Ciao a tutti, appassionati di scienza e misteri del corpo umano! Oggi voglio portarvi con me in un viaggio affascinante nel mondo della genetica, un viaggio che ci ha portato a scoprire qualcosa di inaspettato su una famiglia di sindromi chiamate RASopatie. Preparatevi, perché stiamo per parlare di un gene, ERF, e di come le sue “versioni alternative” possano causare sintomi simili alla sindrome di Noonan, ma con una svolta sorprendente: niente craniosinostosi!
Un Intricato Sentiero Cellulare: Le RASopatie
Prima di tuffarci nel vivo, facciamo un piccolo ripasso. Avete mai sentito parlare della via di segnalazione RAS/MAPK? Immaginatela come una complessa rete di comunicazione all’interno delle nostre cellule, fondamentale per regolare processi vitali come la crescita, la differenziazione e la sopravvivenza cellulare. È un meccanismo delicatissimo.
Quando qualcosa in questa via non funziona a dovere a causa di mutazioni genetiche germinali (cioè presenti fin dal concepimento), possono insorgere delle sindromi dello sviluppo note collettivamente come RASopatie. Tra le più conosciute ci sono la sindrome di Noonan (NS), la sindrome di Costello e la sindrome cardio-facio-cutanea. Queste condizioni condividono spesso caratteristiche come bassa statura, tratti del viso particolari, difetti cardiaci congeniti e ritardi nello sviluppo.
I geni coinvolti sono tanti (più di 20!), tra cui i famosi *PTPN11*, *SOS1*, *KRAS*, *BRAF*, e molti altri. Eppure, nonostante le nostre conoscenze, circa il 20% delle persone con sintomi suggestivi di una RASopatia rimane senza una diagnosi molecolare precisa. Questo ci dice che ci sono ancora geni da scoprire o meccanismi da capire meglio. Ed è qui che entra in gioco il nostro protagonista: il gene ERF.
ERF: Un Repressore con un Doppio Volto?
Il gene ERF (ETS2 repressor factor) codifica per una proteina che fa parte della famiglia dei fattori di trascrizione ETS. Il suo ruolo principale? Agire come un “freno” trascrizionale, spegnendo l’attività di altri geni. ERF è un attore a valle della cascata RAS/MAPK, in particolare viene regolato da ERK1/2.
Finora, le varianti germinali nel gene ERF erano state associate principalmente a una condizione chiamata craniosinostosi correlata a ERF (CRS4), caratterizzata dalla chiusura prematura delle suture craniche, oltre ad anomalie facciali e disturbi dello sviluppo. Si pensava che una ridotta funzione di ERF (aploinsufficienza) portasse a questo quadro clinico.
Ma la biologia ama sorprenderci! Recentemente, sono emerse segnalazioni di varianti di ERF (soprattutto quelle che troncano la proteina) in individui con un fenotipo simil-Noonan, ma senza la craniosinostosi che ci si aspetterebbe. Questo ha aperto un sacco di domande: qual è il vero spettro clinico associato a ERF? Come funzionano esattamente queste varianti?
La Nostra Scoperta: Varianti ERF in Casi Simil-Noonan Senza Craniosinostosi
Ed eccoci al cuore della nostra ricerca. Utilizzando il sequenziamento dell’intero esoma (WES), una tecnica potentissima che legge quasi tutte le regioni codificanti dei nostri geni, abbiamo analizzato il DNA di alcune persone con sospetta sindrome di Noonan per cui le analisi precedenti sui geni noti non avevano dato risultati.
Colpo di scena! In tre individui abbiamo identificato varianti “sospette” proprio nel gene ERF:
- Una variante missenso (p.Gly53Arg – G53R), che causa la sostituzione di un singolo amminoacido nella proteina.
- Due varianti troncanti (p.Arg183* – R183* e p.Gly299Argfs*9 – G299Rfs), che portano alla produzione di una proteina ERF più corta del normale.
Inoltre, riesaminando con la tecnica classica del sequenziamento Sanger un gruppo più ampio di 81 persone con sospetta RASopatia senza diagnosi, abbiamo scovato un’altra variante troncante (p.Lys401Glufs*10 – K401Efs) in un ulteriore individuo.
La cosa davvero interessante? Tutti e quattro questi individui avevano una diagnosi clinica di sindrome simil-Noonan, con caratteristiche facciali tipiche (ipertelorismo, rime palpebrali oblique verso il basso, orecchie a basso impianto e ruotate posteriormente), bassa statura e disturbi dello sviluppo (ritardo del linguaggio, difficoltà di apprendimento). Tre su quattro avevano anche macrocefalia (testa più grande della norma). Ma nessuno di loro presentava craniosinostosi!

Abbiamo anche verificato che le varianti fossero presenti nei genitori, ove possibile. Nel caso della variante missenso G53R, il padre la possedeva e mostrava sintomi lievi (ipertelorismo, macrocefalia, bassa statura). Negli altri due casi analizzati, le varianti troncanti erano de novo, cioè comparse per la prima volta nei figli.
Questi risultati suggeriscono fortemente che le varianti patogeniche in ERF possono causare un quadro clinico che si sovrappone notevolmente alla sindrome di Noonan, ma senza necessariamente coinvolgere la fusione prematura delle suture craniche.
Dentro la Cellula: Cosa Fanno le Varianti ERF Mutate?
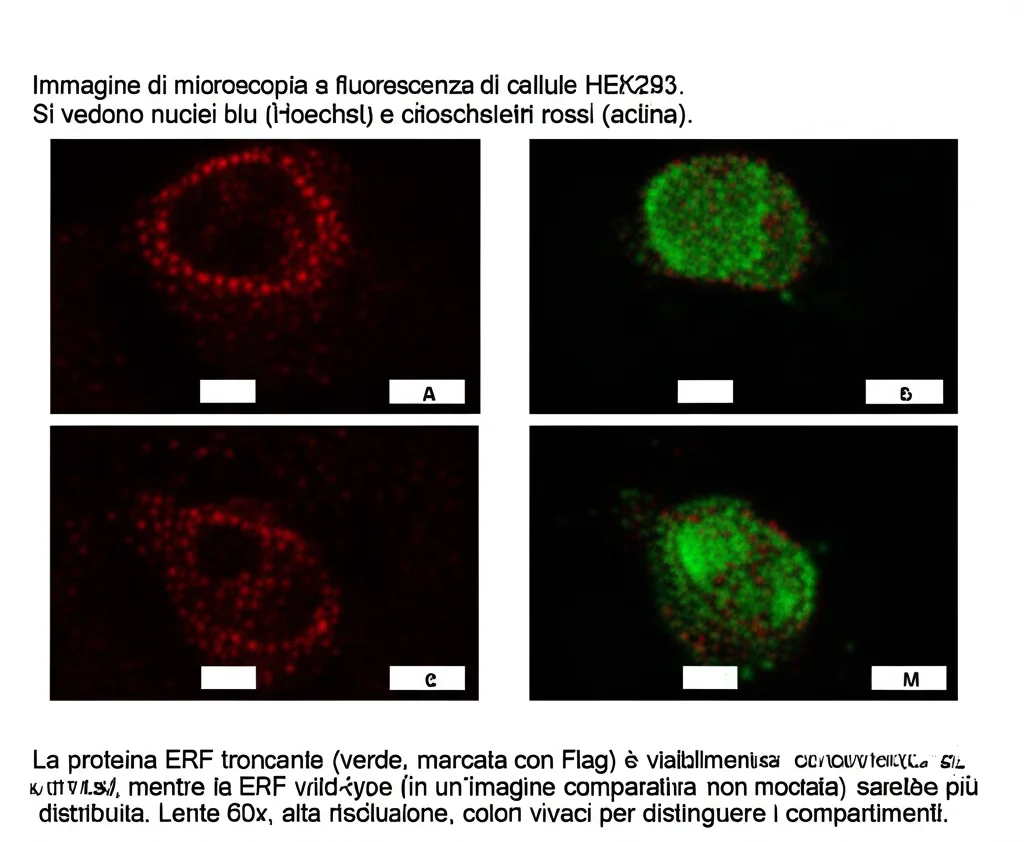
Identificare le varianti è solo il primo passo. La vera sfida è capire *cosa* fanno di diverso rispetto alla proteina normale. Così, ci siamo messi al lavoro in laboratorio, creando versioni della proteina ERF (sia normale, “wild-type”, che mutata) per studiarne il comportamento in cellule coltivate.
Dove va la proteina ERF?
Normalmente, la proteina ERF si trova principalmente nel nucleo cellulare quando la cellula è “a riposo” (senza stimoli esterni). Quando la cellula viene stimolata (ad esempio, con siero), la via RAS/MAPK si attiva, ERK1/2 fosforila ERF, e questa si sposta dal nucleo al citoplasma, perdendo la sua funzione repressiva. È un meccanismo di regolazione finissimo.
Abbiamo osservato che la ERF normale (wild-type) e le varianti missenso (come la G53R che abbiamo trovato, ma anche altre note come R83W) si comportavano più o meno come previsto: si spostavano dal nucleo al citoplasma dopo stimolazione.
Ma le varianti troncanti (R183*, G299Rfs, K401Efs) erano tutta un’altra storia! Queste proteine rimanevano prevalentemente “intrappolate” nel nucleo, anche dopo la stimolazione. Sembra che manchi loro un pezzo fondamentale per poter uscire correttamente dal nucleo. Questa localizzazione anomala potrebbe già essere un indizio importante.
ERF e la crescita cellulare
ERF agisce come un freno. Ci siamo chiesti: le varianti mutate frenano ancora la crescita? Abbiamo usato cellule simili a quelle che formano l’osso (osteoblasti, le MC3T3-E1) e abbiamo visto che la ERF normale rallentava la loro proliferazione. Interessante, le varianti troncanti R183* e G299Rfs avevano perso questa capacità: le cellule che le esprimevano crescevano di più rispetto a quelle con ERF normale! Sembra che il freno sia rotto.
ERF, RUNX2 e la formazione dell’osso
Sappiamo che ERF può interagire con un altro fattore importante per la formazione dell’osso, chiamato RUNX2. RUNX2 promuove la differenziazione degli osteoblasti. A volte, ERF agisce come un antagonista di RUNX2.
Abbiamo usato dei “reporter” a luciferasi (sì, come le lucciole!) per misurare l’attività trascrizionale legata a RUNX2. I risultati? La ERF normale reprimeva questa attività, come ci si aspettava. Ma le varianti troncanti R183* e G299Rfs non solo non reprimevano, ma in alcuni contesti sembravano addirittura *aumentare* l’attività legata a RUNX2! Un altro segnale che qualcosa non va nella loro funzione di controllo.
L’effetto finale: l’ossificazione
Tutti questi indizi puntavano in una direzione: forse queste varianti ERF influenzano direttamente la capacità delle cellule di formare osso. Abbiamo quindi coltivato le nostre cellule MC3T3-E1 in un terreno che induce l’ossificazione e abbiamo misurato due cose:
- L’attività della fosfatasi alcalina (ALP), un enzima precoce dell’ossificazione.
- La mineralizzazione vera e propria, colorando i depositi di calcio con Alizarina Rossa.
I risultati sono stati sorprendenti! La ERF normale riduceva sia l’attività ALP che la mineralizzazione rispetto alle cellule di controllo. Ma le cellule che esprimevano le varianti patogeniche (sia la missenso G53R che le troncanti R183*, G299Rfs, K401Efs) mostravano una maggiore capacità di ossificazione rispetto a quelle con ERF normale. In pratica, la perdita di funzione di ERF sembra togliere un freno al processo di formazione dell’osso in queste cellule.

Cosa Significa Tutto Questo? Un Nuovo Pezzo del Puzzle
Mettiamo insieme i pezzi. Abbiamo trovato varianti ERF (una missenso e diverse troncanti) in persone con un fenotipo simil-Noonan ma senza craniosinostosi. I nostri esperimenti in laboratorio mostrano che queste varianti sono “loss-of-function”, cioè causano una perdita della normale funzione repressiva di ERF. In particolare:
- Le varianti troncanti rimangono bloccate nel nucleo.
- Alcune varianti troncanti perdono la capacità di frenare la crescita cellulare.
- Le varianti troncanti non riescono a reprimere l’attività legata a RUNX2.
- Tutte le varianti patogeniche testate (inclusa la missenso G53R) portano a un aumento dell’ossificazione in cellule osteoblastiche rispetto alla ERF normale.
Questo ci suggerisce un meccanismo affascinante: la perdita di funzione di ERF, un repressore a valle di ERK, potrebbe portare a una sorta di “iperattivazione” indiretta delle vie che promuovono l’ossificazione. Questo potrebbe spiegare le anomalie craniofacciali osservate in questi pazienti, anche in assenza della fusione completa delle suture tipica della craniosinostosi. Forse una crescita o chiusura disregolata in punti specifici delle suture del cranio e della faccia contribuisce ai tratti Noonan-like.
È anche interessante notare che, nei nostri esperimenti, le varianti troncanti sembravano avere effetti funzionali più marcati rispetto alla variante missenso G53R (tranne che per l’ossificazione, dove tutte le mutazioni patogene davano un effetto simile). Questo si allinea con l’idea, emersa anche dalla letteratura, che le varianti troncanti potrebbero essere associate a fenotipi clinicamente più severi.
Conclusioni (Provvisorie) di un Viaggio Incredibile
Questo studio aggiunge un tassello importante alla nostra comprensione delle RASopatie e delle sindromi correlate. Ci dice che le varianti ERF non causano solo craniosinostosi, ma possono anche portare a un fenotipo simil-Noonan senza fusione delle suture. La perdita della funzione repressiva di ERF, specialmente per quanto riguarda il controllo dell’ossificazione, sembra essere la chiave.
È possibile che la perdita di funzione di questo repressore a valle di ERK possa, di fatto, potenziare la segnalazione della via RAS/MAPK, portando a un fenotipo simil-Noonan attraverso un meccanismo comune ad altre RASopatie.
Certo, la ricerca non finisce qui. Bisogna capire meglio perché alcuni individui con varianti ERF sviluppano craniosinostosi e altri no, e quali altri fattori (genetici o ambientali) modulano il fenotipo. Ma ogni scoperta come questa ci avvicina a una comprensione più profonda di queste complesse condizioni genetiche e, speriamo, a future strategie terapeutiche. È la bellezza della scienza: un mistero risolto apre le porte a mille nuove domande!
Fonte: Springer





