TLR2: La Proteina “Canaglia” che Fa Squadra con Diabete e Arterie Intasate
Ciao a tutti! Oggi voglio parlarvi di una scoperta che mi ha davvero incuriosito, una di quelle che ti fa dire “Wow, la biologia è pazzesca!”. Immaginatevi il nostro corpo come una metropoli super complessa, con un sacco di meccanismi che devono funzionare alla perfezione. A volte, però, qualche “cittadino” molecolare decide di fare il doppio gioco, e le cose iniziano ad andare storte. È un po’ quello che succede quando il diabete mellito si mette d’accordo con l’aterosclerosi, quella brutta bestia che ci indurisce e ostruisce le arterie. Un duo davvero pericoloso, che purtroppo colpisce tantissime persone.
Ma cosa scatena questa alleanza nefasta? Beh, i ricercatori sono sempre a caccia dei colpevoli, e uno dei sospettati principali, emerso da uno studio recente, si chiama Toll-like receptor-2, o più semplicemente TLR2. Sembra un nome da robot di un film di fantascienza, vero? In realtà, è una proteina che di solito fa parte del nostro sistema di difesa, una specie di sentinella. Il problema è che, a volte, questa sentinella può diventare un po’ troppo zelante, o peggio, passare dalla parte dei “cattivi”.
Ma cos’è esattamente questa “DMA”?
Prima di addentrarci nei meandri della ricerca, facciamo un piccolo ripasso. Il diabete mellito, come saprete, è quel disturbo metabolico in cui il corpo non produce abbastanza insulina o non la usa come dovrebbe. Risultato? Zuccheri alle stelle nel sangue e, a lungo andare, un sacco di organi che iniziano a soffrire. L’aterosclerosi, invece, è un’infiammazione cronica che fa ispessire le pareti delle arterie e le riempie di depositi di grasso. Quando queste due condizioni si presentano insieme, parliamo di aterosclerosi associata al diabete mellito (DMA). È una delle complicanze vascolari più critiche e, purtroppo, le terapie attuali, come farmaci per abbassare la glicemia o anticoagulanti, spesso non bastano. Ecco perché scovare nuovi bersagli terapeutici è fondamentale.
TLR2: L’indiziato speciale sotto la lente d’ingrandimento
Torniamo al nostro TLR2. Questa proteina appartiene a una famiglia di recettori (i Toll-like receptors, appunto) che sono cruciali per il nostro sistema immunitario innato, la prima linea di difesa contro microbi e invasori. Quando un TLR si attiva, scatena una cascata di segnali che portano alla produzione di citochine e altre molecole infiammatorie. Utile, certo, ma se il processo sfugge di mano, l’infiammazione diventa cronica e dannosa.
Nello studio che ho letto, i ricercatori hanno voluto vederci chiaro sul ruolo di TLR2 nella DMA. Hanno preso campioni da 30 pazienti con DMA e 30 persone sane, e indovinate un po’? I livelli di TLR2 erano significativamente più alti nei pazienti con DMA! Non solo, ma hanno anche creato un modello di DMA in laboratorio, usando cellule endoteliali microvascolari cardiache umane (HCMEC), trattandole con tanto glucosio e lipoproteine ossidate (quelle “cattive” che contribuiscono all’aterosclerosi). Anche in questo modello cellulare, TLR2 era su di giri.
Ma cosa combinava questo TLR2 “impazzito”? Beh, le cellule del modello DMA mostravano una vitalità ridotta e un aumento della piroptosi. La piroptosi è una forma particolare di morte cellulare programmata che scatena un’intensa infiammazione, rilasciando citochine pro-infiammatorie come l’interleuchina-1β (IL-1β) e l’interleuchina-18 (IL-18). E infatti, anche i livelli di queste citochine e delle proteine legate alla piroptosi (come ASC, NLRP3 e GSDMD-N) erano più alti. Sembra proprio che TLR2 stesse gettando benzina sul fuoco dell’infiammazione!

Il meccanismo d’azione: come TLR2 orchestra il danno
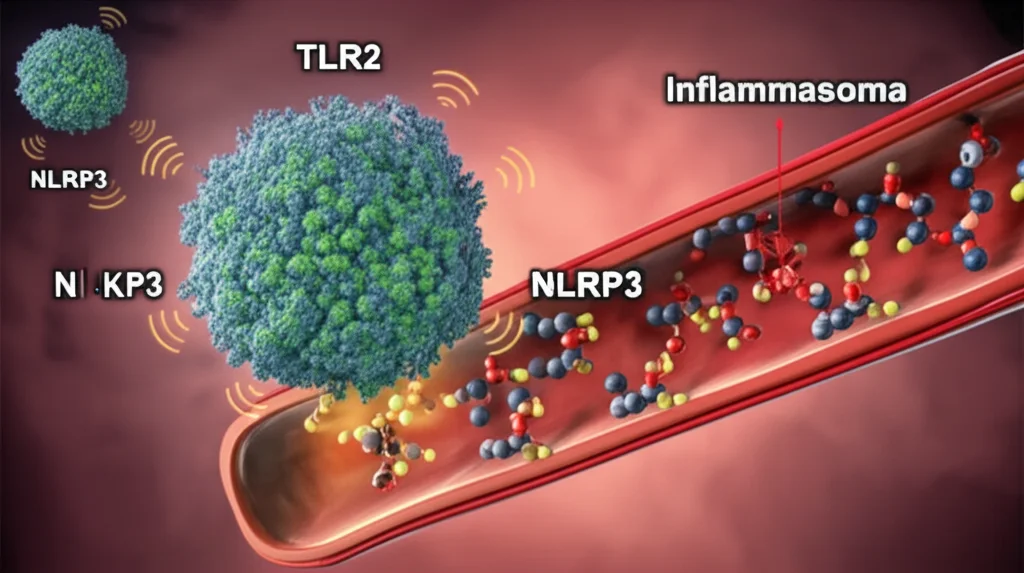
Ok, TLR2 è alto e fa danni, ma come? Qui la storia si fa ancora più interessante. Sembra che TLR2 non agisca da solo, ma attivi due complici principali: l’inflammasoma NLRP3 e la via di segnalazione MyD88/NF-κB.
L’inflammasoma NLRP3 è un complesso multiproteico che, una volta attivato, è responsabile proprio della maturazione e del rilascio di IL-1β e IL-18, le citochine che abbiamo visto prima. È come un interruttore generale dell’infiammazione. Lo studio ha dimostrato che l’attivazione di TLR2 accendeva questo interruttore.
L’altra via coinvolta è quella di MyD88/NF-κB. MyD88 è una proteina adattatrice che si lega a TLR2 quando questo viene attivato. Questo legame dà il via a una serie di reazioni a catena che culminano nell’attivazione di NF-κB. NF-κB è un fattore di trascrizione, una specie di “capo operaio” che entra nel nucleo della cellula e ordina la produzione di geni coinvolti nell’infiammazione, nella sopravvivenza cellulare e in molte altre funzioni. I ricercatori hanno visto che TLR2 interagisce fisicamente con MyD88 e ne regola la stabilità attraverso un processo chiamato ubiquitinazione (una sorta di “etichettatura” delle proteine per la degradazione o per altre funzioni). In pratica, TLR2 teneva MyD88 bello attivo, e di conseguenza anche NF-κB era costantemente allertato, pronto a fomentare l’infiammazione.
Spegnere TLR2: una possibile soluzione?
La prova del nove, in questi casi, è vedere cosa succede se si “spegne” il sospettato. Ebbene, quando i ricercatori hanno inibito TLR2 nel loro modello cellulare di DMA, le cose sono migliorate parecchio! La vitalità cellulare è aumentata, la piroptosi si è ridotta, i livelli delle proteine infiammatorie e delle citochine IL-1β e IL-18 sono calati. Non solo, ma l’inibizione di TLR2 ha bloccato efficacemente l’attivazione della via MyD88/NF-κB.
Al contrario, quando hanno “iperattivato” TLR2, tutti gli effetti negativi sono peggiorati: meno vitalità cellulare, più piroptosi e via MyD88/NF-κB a mille. Però, se in queste condizioni bloccavano NF-κB con un inibitore specifico (chiamato BAY 11-7082), gli effetti deleteri della sovraespressione di TLR2 venivano contrastati. Questo conferma che NF-κB è un attore chiave a valle di TLR2.
Questi risultati non si sono limitati alle piastrine di Petri. Lo studio ha incluso anche un modello animale, dei topolini diabetici con aterosclerosi. Anche in questi topolini, “silenziare” TLR2 ha portato a una riduzione dell’infiammazione e dell’aterosclerosi, con una minor deposizione di lipidi nelle loro aorte. Immaginate le loro arterie che tirano un sospiro di sollievo!

Cosa ci portiamo a casa da questa ricerca?
Beh, per me è chiaro: TLR2 sembra essere un protagonista di primo piano nella progressione del diabete associato ad aterosclerosi. Agisce come un interruttore che, una volta acceso, scatena l’inferno infiammatorio attraverso l’attivazione dell’inflammasoma NLRP3 e della via MyD88/NF-κB. È come se TLR2 fosse il mandante, e NLRP3 e MyD88/NF-κB i suoi scagnozzi più fidati.
La cosa entusiasmante è che questi risultati aprono la porta a nuove possibili strategie terapeutiche. Se riusciamo a trovare un modo per “calmare” TLR2 o per bloccare le vie che attiva, potremmo avere una nuova arma per combattere la DMA. Certo, la strada è ancora lunga. Come sottolineano gli stessi autori, lo studio ha alcune limitazioni, come il numero relativamente piccolo di pazienti e il fatto che i modelli di laboratorio, per quanto utili, non replicano mai al 100% la complessità del corpo umano.
Tuttavia, la direzione sembra promettente. Si potrebbe pensare allo sviluppo di piccole molecole che inibiscano specificamente TLR2, o a terapie combinate che colpiscano sia TLR2 sia i suoi “complici” a valle. Chissà, magari un giorno avremo farmaci capaci di disinnescare questa proteina “canaglia” e proteggere le arterie dei pazienti diabetici.
Io, da appassionato, non vedo l’ora di scoprire i prossimi capitoli di questa avvincente storia scientifica. E voi?
Fonte: Springer