Cancro Ovarico Resistente? Ho Trovato il Tallone d’Achille: Bersagliare AKT1!
Ciao a tutti! Oggi voglio parlarvi di una sfida enorme nella lotta contro i tumori, una di quelle che ci tiene svegli la notte in laboratorio: il cancro ovarico. Sapete, è una delle forme più letali di tumore ginecologico, e il motivo principale è che spesso diventa resistente alle chemioterapie e tende a ripresentarsi. Un vero osso duro.
Recentemente, abbiamo notato un legame sempre più forte tra questo problema e una proteina chiamata SOX2. SOX2 è un fattore di trascrizione, una specie di “interruttore” genetico, fondamentale per le cellule staminali. Il problema è che quando si esprime troppo nelle cellule tumorali ovariche, sembra renderle più resistenti ai farmaci e peggiorare la prognosi delle pazienti. Immaginate SOX2 come un generale che ordina alle cellule tumorali di resistere all’attacco della chemio.
Il Mistero di SOX2: Perché è Così Abbondante?
Studiando diverse linee cellulari di cancro ovarico e campioni di tumori reali, abbiamo confermato quello che già si sospettava: SOX2 è spesso presente in quantità eccessive. Ma la domanda era: perché? A volte, un aumento di proteina è dovuto a più copie del gene che la produce (amplificazione genica) o a una maggiore “lettura” di quel gene (trascrizione). Ma analizzando i dati, anche quelli di grandi database come il TCGA, abbiamo visto che l’amplificazione del gene SOX2 o un aumento della sua trascrizione spiegavano solo una piccola parte dei casi. C’era qualcos’altro sotto.
Abbiamo confrontato i livelli di SOX2 (proteina, mRNA e copie del gene) in diverse linee cellulari e abbiamo visto che non c’era una correlazione diretta e forte tra le copie del gene e la quantità di proteina finale. Anche l’analisi dei dati TCGA ha mostrato che solo una piccola percentuale di tumori ovarici aveva sia l’amplificazione del gene SOX2 sia alti livelli del suo mRNA. Eppure, la proteina SOX2 era presente in oltre il 60-70% dei tumori analizzati, sia nel nostro studio su campioni di pazienti (abbiamo usato una tecnica chiamata tissue microarray) sia in studi precedenti. Questo ci ha fatto pensare: forse il problema non è tanto *quanto* SOX2 viene prodotto, ma quanto a lungo riesce a sopravvivere nella cellula prima di essere eliminato. Doveva esserci un meccanismo di regolazione “post-trascrizionale” in gioco, qualcosa che stabilizzasse la proteina.
A Caccia del Regista Occulto: Lo Screening degli Inibitori
Se SOX2 è così importante per la resistenza e la “cattiveria” del tumore, ma è difficile da colpire direttamente con farmaci (è un fattore di trascrizione, notoriamente “undruggable”), allora forse potevamo trovare chi lo controlla e colpire quello? Abbiamo ipotizzato che qualche via di segnalazione cellulare, magari iperattiva nel cancro ovarico, potesse essere responsabile della stabilizzazione di SOX2.
Così, abbiamo messo alla prova un arsenale di piccole molecole, degli inibitori di chinasi. Le chinasi sono enzimi che aggiungono gruppi fosfato ad altre proteine, modificandone l’attività o la stabilità. Ne abbiamo selezionati 58, mirati contro 30 delle principali chinasi cellulari implicate nel cancro. Abbiamo trattato le cellule tumorali ovariche (in particolare le Pa-1, che esprimono molto SOX2) con questi inibitori e abbiamo guardato cosa succedeva ai livelli di proteina SOX2.
I risultati sono stati illuminanti! Tra i candidati più efficaci nel ridurre SOX2 c’erano inibitori di:
- Un gruppo di chinasi tra cui PKA, PKG, MLCK e PKC (la PKC era già stata collegata a SOX2).
- La chinasi del recettore del fattore di crescita epidermico (EGFRK), anche questa nota per influenzare SOX2.
- E, soprattutto, gli inibitori di AKT!
Dato che gli inibitori di AKT erano tra i più potenti e che AKT è una chinasi spesso iperattiva in molti tumori, inclusi quelli ovarici, abbiamo deciso di concentrarci su di lei. In particolare, su AKT1.
AKT1: Il Burattinaio di SOX2
Abbiamo quindi verificato: l’inibizione specifica di AKT1 con un farmaco sperimentale chiamato MK2206 riduceva davvero SOX2 nelle cellule di cancro ovarico (linee Pa-1 e OVCAR3)? La risposta è stata un sonoro sì! E l’effetto era dose-dipendente: più inibitore usavamo, meno SOX2 restava. MK2206 bloccava l’attività di AKT1, come dimostrato dalla riduzione della sua forma fosforilata (attiva).
Ma come faceva AKT1 a controllare SOX2? Abbiamo visto che MK2206 riduceva un po’ anche l’mRNA di SOX2, quindi un certo controllo sulla trascrizione c’è. Tuttavia, l’effetto principale sembrava essere sulla stabilità della proteina. Infatti, se bloccavamo il sistema di smaltimento delle proteine della cellula (il proteasoma) con un altro farmaco (MG132), l’effetto di MK2206 su SOX2 veniva annullato. Questo significa che AKT1 protegge SOX2 dalla degradazione via proteasoma.
Per confermarlo, abbiamo misurato “l’emivita” di SOX2, cioè quanto tempo impiega la cellula a dimezzarne la quantità. Usando la cicloesimide (CHX), che blocca la produzione di nuove proteine, abbiamo visto che in condizioni normali SOX2 durava circa 10 ore. Ma trattando le cellule con MK2206, l’emivita crollava a meno di 4 ore! AKT1, quindi, agisce come una guardia del corpo per SOX2.
Ma come fa esattamente? Lavori precedenti, anche nostri, avevano mostrato che AKT1 può fosforilare SOX2 (cioè attaccargli un gruppo fosfato) su un aminoacido specifico, la treonina 116 (T116). Questa fosforilazione sembrava proteggerlo. Abbiamo verificato se accadesse anche nel cancro ovarico: sì! Esprimendo più AKT1 nelle cellule, aumentava la quantità di SOX2 fosforilato in T116.
Per la prova del nove, abbiamo creato delle versioni mutanti di SOX2: una in cui T116 non poteva essere fosforilata (T116A) e una che mimava la fosforilazione costante (T116D). Abbiamo inserito queste versioni mutanti nelle cellule OVCAR3. Poi abbiamo trattato le cellule con MK2206. Risultato? L’inibitore riduceva i livelli del SOX2 “normale” endogeno, ma non aveva effetto sui livelli delle due forme mutanti (T116A e T116D)! Questo dimostra che la fosforilazione di T116 è proprio il meccanismo chiave con cui AKT1 stabilizza SOX2.
Colpire AKT1 per Fermare il Cancro (e la sua Resistenza)
Ok, abbiamo capito il meccanismo. Ma quali sono le implicazioni pratiche? SOX2 è legato alla capacità delle cellule tumorali di proliferare, formare colonie e comportarsi come cellule staminali tumorali (quelle più resistenti e capaci di dare metastasi e recidive). Abbiamo quindi verificato se bloccando SOX2 o AKT1 si ottenessero effetti simili.
Effettivamente, riducendo SOX2 con tecniche di silenziamento genico (shRNA), abbiamo visto che le cellule Pa-1 e OVCAR3 proliferavano meno, formavano meno colonie e avevano una ridotta capacità di formare “sfere tumorali” (un test per misurare la staminalità). Ebbene, facendo la stessa cosa con AKT1 (silenziandolo con shRNA o inibendolo con MK2206), abbiamo ottenuto risultati praticamente sovrapponibili! Inibire AKT1 frenava la crescita e la “staminalità” delle cellule tumorali SOX2-positive.
La Svolta Terapeutica: Rendere Efficace la Chemioterapia al Platino
E qui arriva la parte più entusiasmante. SOX2 è implicato nella resistenza ai farmaci chemioterapici a base di platino (come cisplatino e carboplatino), che sono lo standard di cura per il cancro ovarico. Potevamo usare l’inibitore di AKT1, MK2206, per rendere queste cellule resistenti di nuovo sensibili alla chemio?
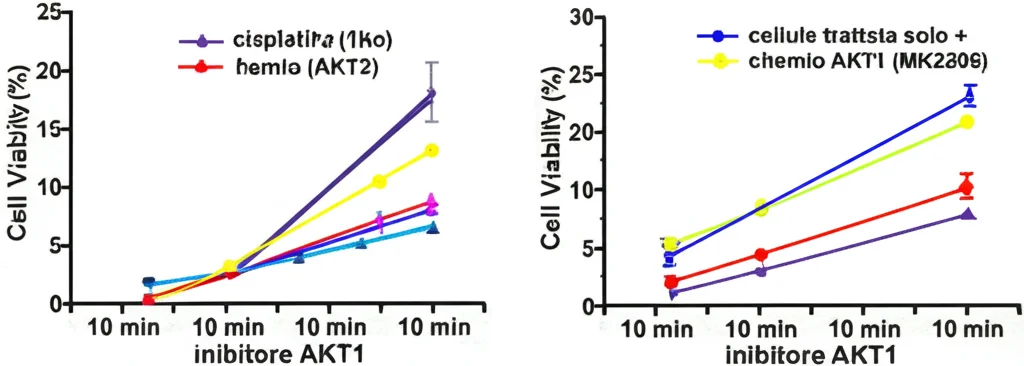
Abbiamo fatto l’esperimento: silenziando SOX2, le cellule diventavano più sensibili al cisplatino (serviva una dose minore per ucciderle, l’IC50 si riduceva). Poi abbiamo provato MK2206. Da solo, inibiva la crescita cellulare, ma la cosa interessante è stata la combinazione. Usando MK2206 insieme a cisplatino o carboplatino, abbiamo osservato un effetto sinergico! Significa che l’effetto combinato era maggiore della somma degli effetti dei singoli farmaci. L’indice di combinazione (CI) era inferiore a 1, indicando sinergia.
La combinazione di MK2206 (a dosi relativamente basse) con cisplatino o carboplatino era molto più efficace nell’inibire la proliferazione e la formazione di colonie delle cellule Pa-1 e OVCAR3 rispetto ai singoli farmaci. E c’è di più: abbiamo notato che la combinazione di MK2206 e carboplatino riduceva i livelli di SOX2 ancora di più rispetto ai singoli trattamenti. Questo suggerisce che parte dell’effetto sinergico potrebbe derivare proprio da un attacco combinato ai livelli di SOX2.
Cosa Significa Tutto Questo? Prospettive Future
In sintesi, il nostro studio suggerisce che nel cancro ovarico, l’eccesso di SOX2 non dipende tanto da anomalie genetiche dirette, quanto da meccanismi che ne aumentano la stabilità, e un attore chiave in questo processo è la chinasi AKT1, che lo protegge dalla degradazione fosforilandolo in T116.
La buona notizia è che, mentre SOX2 è un bersaglio difficile, AKT1 non lo è! Esistono già inibitori di AKT, alcuni dei quali sono in sperimentazione clinica. Il nostro lavoro suggerisce fortemente che inibire AKT1 potrebbe essere una strategia terapeutica promettente per i tumori ovarici SOX2-positivi, specialmente quelli resistenti ai farmaci a base di platino. La combinazione di un inibitore di AKT con la chemioterapia standard potrebbe superare la resistenza e migliorare l’efficacia del trattamento.
Certo, la strada è ancora lunga. Bisogna confermare questi risultati in modelli animali e poi, si spera, nell’uomo. Bisogna anche considerare i potenziali effetti collaterali, visto che SOX2 è importante anche per le cellule staminali normali. Ma aver identificato AKT1 come il “tallone d’Achille” che controlla SOX2 in questo contesto ci apre una nuova, eccitante via da esplorare nella lotta contro questo nemico difficile. Incrociamo le dita!
Fonte: Springer