Sindrome di Claes-Jensen in Palestina: Viaggio nel Cuore di una Diagnosi Genetica Complessa
Ciao a tutti! Mettetevi comodi perché oggi vi porto con me alla scoperta di una storia scientifica davvero particolare, che ci arriva direttamente dalla Palestina. Parleremo di una condizione genetica rara, la sindrome di Claes-Jensen (CJS), e di come un caso specifico abbia acceso i riflettori sulle sfide diagnostiche e sulle incredibili intuizioni che la genetica moderna ci può offrire. Pensate che si tratta del primo caso de novo (cioè una mutazione genetica nuova, non ereditata dai genitori) riportato in Palestina! Una vera e propria pietra miliare.
Cos’è esattamente la Sindrome di Claes-Jensen?
Allora, cerchiamo di capire di cosa stiamo parlando. La sindrome di Claes-Jensen è una forma rara di disabilità intellettiva legata al cromosoma X. Questo significa che il “colpevole” è un gene che si trova su quel cromosoma. Il gene in questione si chiama KDM5C. Immaginatelo come un piccolo operaio specializzato nel nostro corpo: il suo compito è produrre un enzima (una demetilasi istone specifica per la lisina 5C, per i più tecnici) che gioca un ruolo cruciale nella rimodellazione della cromatina e nello sviluppo neurologico. In parole povere, aiuta a regolare l’espressione di altri geni, assicurandosi che tutto funzioni a dovere durante lo sviluppo del cervello. Se questo gene ha una mutazione, beh, le cose possono complicarsi.
Generalmente, i maschi che hanno una mutazione emizigote (cioè hanno una sola copia del cromosoma X, e quindi del gene) nel KDM5C presentano disabilità intellettiva, caratteristiche somatiche particolari (dismorfismi) e ritardi nello sviluppo neurologico. Le mutazioni possono essere trasmesse dalla madre (che di solito è portatrice sana o con sintomi lievi) oppure, come nel caso che stiamo per esplorare, possono essere de novo, cioè comparire spontaneamente nel bambino. Queste mutazioni sono responsabili di una piccola ma significativa percentuale (0.7-2.8%) delle disabilità intellettive legate al cromosoma X.
Un caso palestinese che fa storia
Ed eccoci al cuore della nostra storia. Il protagonista è un bambino palestinese di 2 anni e 10 mesi. La sua vicenda clinica inizia a farsi preoccupante intorno ai 22 mesi, quando, a seguito di una malattia virale acuta, manifesta una regressione dello sviluppo. Immaginate lo smarrimento: un bambino che aveva iniziato a camminare, improvvisamente perde questa capacità. A questo si aggiungono ritardi nello sviluppo già preesistenti e un valore persistentemente elevato di acido lattico nel sangue, che inizialmente ha fatto pensare i medici a un disturbo metabolico.
La gravidanza della madre era stata spontanea, ma con qualche complicazione: un’infezione da COVID-19 al settimo mese e ipertensione nelle fasi finali. Il bambino è nato a 38 settimane con taglio cesareo, proprio a causa dell’ipertensione materna, con un peso di 3700g e senza problemi neonatali immediati. Tuttavia, fin da piccolo, le tappe dello sviluppo erano state raggiunte con ritardo: si è seduto senza supporto a 15 mesi, ha camminato a 18 (per poi perdere questa abilità), e a 24 mesi trasferiva oggetti tra le mani e usava una presa a due dita, pronunciando solo poche semplici parole come “Ma”, “Da”, “Mama” e “Dada”.
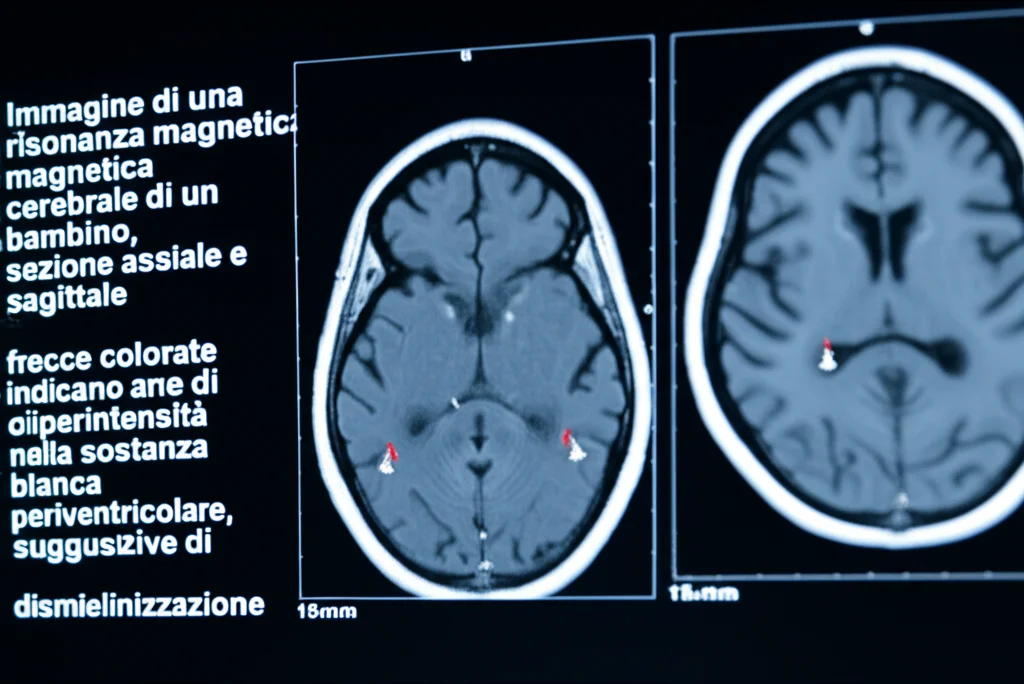
All’esame fisico, non presentava dismorfismi evidenti, ma a livello neurologico si notava un lieve aumento del tono muscolare alle caviglie, iperreflessia (riflessi tendinei profondi vivaci) e un tono assiale normale. Gli esami di laboratorio, oltre all’acido lattico elevato, mostravano globuli rossi microcitici. L’ecocardiogramma era normale, ma la risonanza magnetica cerebrale (MRI) ha rivelato delle lievi iperintensità nella sostanza bianca periventricolare posteriore, un segnale che poteva suggerire problemi di mielinizzazione (la guaina che ricopre le fibre nervose), ma senza anomalie strutturali evidenti.
Il vero punto di svolta è arrivato con l’analisi genetica. È stata eseguita una sequenza dell’intero esoma (WES) basata su un trio (cioè analizzando il DNA del bambino e di entrambi i genitori). Ed ecco la scoperta: una mutazione de novo nel gene KDM5C (specificamente c.2827 C > T p.Arg943*). Questa variante, che causa un codone di stop prematuro, è stata classificata come probabilmente patogenetica secondo le linee guida ACMG/AMP 2021, confermando la diagnosi di sindrome di Claes-Jensen.
Le sfide diagnostiche e il potere della genetica
Questo caso è emblematico per diverse ragioni. Innanzitutto, mette in luce le sfide diagnostiche che si possono incontrare con sindromi così rare. La regressione dello sviluppo dopo un’infezione virale e l’acido lattico elevato potevano facilmente depistare i medici. L’infezione virale, infatti, potrebbe aver agito come uno stress fisiologico che ha esacerbato la presentazione clinica di un disturbo neurogenetico sottostante, ma non c’è prova di un legame causale diretto con la sindrome di Claes-Jensen legata a KDM5C. È più probabile che la regressione rifletta il corso naturale della malattia, e che la coincidenza temporale con l’infezione abbia complicato il processo diagnostico.
In secondo luogo, sottolinea l’importanza cruciale dei test genetici, in particolare del WES, nei disturbi del neurosviluppo. Negli ultimi anni, siamo passati da test come l’Array-CGH (Chromosomal Microarray Analysis), con rese diagnostiche del 16-28%, a tecniche come il WES e il sequenziamento dell’intero genoma (WGS), che offrono rese diagnostiche molto più elevate (50-70% nei casi di disabilità intellettiva grave) e sono ora raccomandati come test di prima o seconda linea. Nel nostro caso, il WES è stato risolutivo.
È interessante notare come alcune caratteristiche cliniche – il ritardo dello sviluppo, la bassa statura (anche se non specificata come marcata nel testo, è tipica della sindrome), l’acido lattico elevato e l’aumento del tono muscolare – abbiano fornito indizi fenotipici che hanno aiutato a interpretare i risultati genetici. Questo ci ricorda quanto sia fondamentale il cosiddetto deep phenotyping, cioè una caratterizzazione clinica approfondita del paziente, per complementare i dati genetici.
La mutazione identificata nel paziente (p.Arg943*) è una variante nonsenso che porta a un codone di stop prematuro, ed è stata considerata “likely pathogenic” sulla base di criteri solidi:
- PVS1 (molto forte): È una variante null (nonsenso) in un gene dove la perdita di funzione è un meccanismo di malattia ben stabilito.
- PM2 (moderato): Assente nei database di popolazione come gnomAD ed ExAC.
- PP5 (supportivo): Già riportata in un paziente con fenotipo sovrapponibile.
Inoltre, analisi in silico (cioè al computer) come MutationTaster prevedevano che la variante fosse patogenetica, e la sua classificazione era supportata dall’alta intolleranza del gene alla perdita di funzione. Il fatto che non sia stata rilevata nella madre ha confermato la sua origine de novo.

Le alterazioni della sostanza bianca osservate alla MRI, suggestive di demielinizzazione, sono in linea con il ruolo noto del KDM5C nello sviluppo cerebrale e supportano ulteriormente il legame tra anomalie strutturali del cervello e alterazioni della sostanza bianca negli individui con mutazioni in questo gene.
Cosa ci insegna questo caso?
Questo caso palestinese non è solo un numero in più nelle statistiche. È un contributo prezioso alla letteratura scientifica, ancora limitata, su questa condizione. Ci ricorda che la sindrome di Claes-Jensen, seppur rara, e ancor più rara nelle sue varianti de novo, deve essere considerata nella diagnosi differenziale dei sintomi del neurosviluppo. Un riconoscimento precoce è fondamentale per una gestione accurata e per migliorare la comprensione clinica della malattia.
Purtroppo, ad oggi, non esistono terapie mirate per le disabilità intellettive legate alle mutazioni del KDM5C. La gestione rimane principalmente sintomatica, volta ad alleviare i sintomi e a supportare il paziente e la famiglia. Tuttavia, ogni nuovo caso documentato, ogni nuova intuizione genetica, ci avvicina un po’ di più alla comprensione profonda di questi meccanismi complessi. E chissà, forse un giorno, anche a possibili strategie terapeutiche.
Insomma, questa storia ci dimostra ancora una volta quanto sia affascinante e complesso il mondo della genetica e quanto sia importante la ricerca, soprattutto per quelle malattie rare che colpiscono poche persone, ma che per quelle persone e le loro famiglie rappresentano un intero universo di sfide e speranze. Aumentare la consapevolezza tra i clinici è il primo passo per non lasciare indietro nessuno.
Fonte: Springer