Cellule Tumorali Addormentate? Non Proprio: La Senescenza è la Nuova Resistenza ai Farmaci nel Cancro al Seno!
Ciao a tutti! Oggi voglio parlarvi di qualcosa che mi ha davvero fatto riflettere ultimamente, un meccanismo affascinante e un po’ subdolo che le cellule tumorali, in particolare quelle del cancro al seno, mettono in atto per sopravvivere alle terapie. Parliamo della senescenza indotta da terapia (TIS).
Per anni, abbiamo pensato alla senescenza – uno stato in cui le cellule smettono di dividersi – come a un risultato positivo della chemioterapia. L’idea era: se non possiamo uccidere tutte le cellule tumorali, almeno possiamo “addormentarle” permanentemente, bloccando la loro crescita. Sembrava una buona strategia, no? Beh, forse ci siamo illusi un po’.
La Sorpresa: La Senescenza Non è Così Permanente
Quello che abbiamo scoperto, lavorando con diverse linee cellulari di cancro al seno (MCF7, T47D, MDA-MB-231, Hs578T – nomi tecnici, ma rappresentano diversi tipi di tumore al seno), è che questa senescenza indotta dai farmaci (nel nostro caso, la doxorubicina, un comune chemioterapico) non è affatto permanente.
Abbiamo trattato queste cellule con dosi massicce di farmaco, abbastanza da ucciderne oltre il 90%. Le poche sopravvissute entravano in uno stato che sembrava proprio senescenza: diventavano più grandi, piatte, smettevano di dividersi e mostravano tutti i marcatori classici, come la positività alla colorazione SA-β-Gal, l’aumento della proteina p21 (un freno del ciclo cellulare) e la diminuzione di LMNB1 (un componente della membrana nucleare). Avevano anche danni al DNA, più mitocondri e lisosomi… insomma, sembravano proprio cellule “vecchie” e fuori gioco.
E qui arriva la sorpresa: dopo alcune settimane (parliamo di più di 30 giorni!), una piccolissima frazione di queste cellule “dormienti” (circa 1 su 10 milioni o anche meno) riusciva a risvegliarsi! Ricominciavano a proliferare come se nulla fosse, perdendo tutte le caratteristiche della senescenza. Abbiamo chiamato queste cellule “REPOP” (ripopolanti). Questo fenomeno si è verificato in tutte le linee cellulari testate. Accidenti! La senescenza, quindi, non era la fine della storia, ma solo una pausa strategica.
Un Meccanismo di Difesa Inaspettato: La Resistenza ai Farmaci
Ma la cosa ancora più preoccupante è venuta fuori quando abbiamo provato a trattare nuovamente queste cellule senescenti (TIS). Se somministravamo una seconda dose di doxorubicina alle cellule TIS (prima che si risvegliassero), queste mostravano una resistenza significativa. Incredibile! Lo stato di senescenza le proteggeva dal farmaco. E la cosa si ripeteva: se prendevamo le cellule REPOP (quelle risvegliate) e le trattavamo di nuovo con doxorubicina, rientravano in senescenza (le abbiamo chiamate re-TIS) e ridiventavano resistenti!
A questo punto, ci siamo chiesti: questa resistenza vale solo per la doxorubicina o è più generale? Abbiamo testato ben 63 farmaci antitumorali approvati dalla FDA, con meccanismi d’azione diversissimi. I risultati sono stati sbalorditivi: le cellule TIS erano resistenti a circa metà di questi farmaci!
- Resistenza comune agli antimetaboliti (come gemcitabina, citarabina, clofarabina…).
- Inefficacia di alcuni inibitori della topoisomerasi II (mitoxantrone, pixantrone) e di agenti alchilanti (clormetina, melfalan).
- Resistenza a inibitori specifici come pevonedistat (neddilazione) e tipifarnib (farnesiltransferasi).
La cosa strana è che questa resistenza non dipendeva solo dal fatto che le cellule non si dividevano. Alcuni farmaci che colpiscono cellule in rapida divisione (come AT7519 o ibrutinib) riuscivano comunque a uccidere le cellule TIS, mentre altri, che dovrebbero funzionare anche su cellule quiescenti (come carfilzomib), erano inefficaci. Un profilo di resistenza/sensibilità davvero unico e non spiegabile facilmente con la sola mancanza di proliferazione. Pensate che farmaci comunissimi nel trattamento del cancro al seno come paclitaxel e docetaxel (taxani) erano efficaci sulle cellule normali (CTR) e su quelle ripopolate (REPOP), ma totalmente inutili contro le cellule TIS e re-TIS.
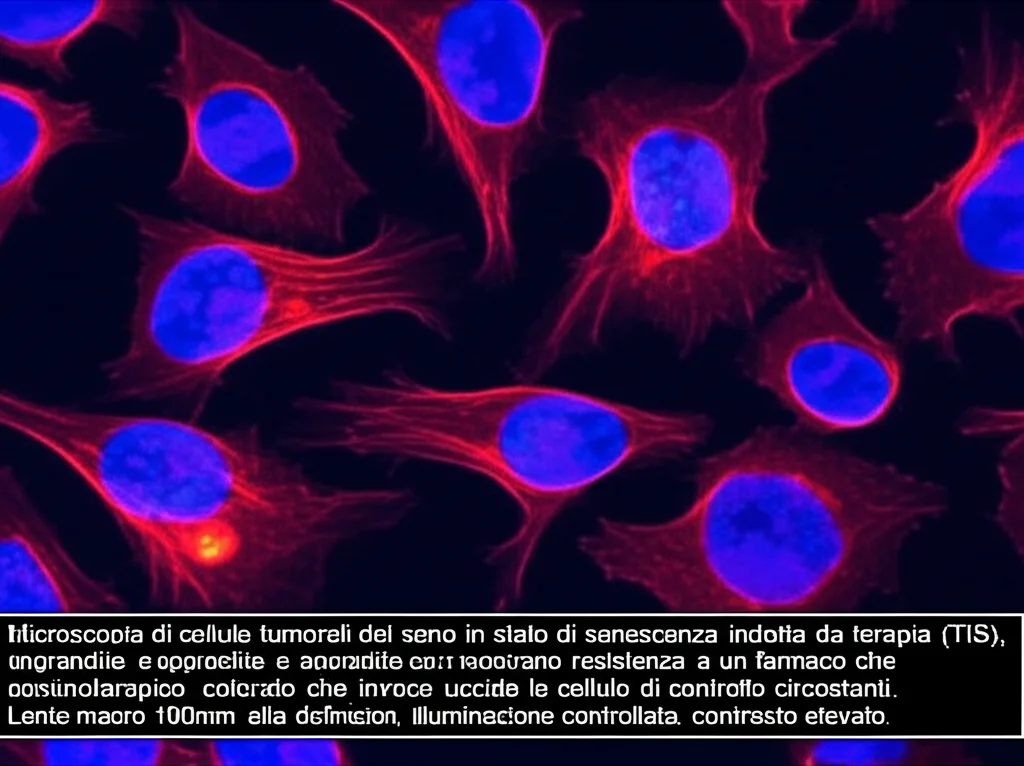
Cercando di Eliminare le Cellule “Addormentate”: Il Fallimento dei Senolitici
Negli ultimi anni si è parlato molto di “senolitici”, farmaci progettati per uccidere selettivamente le cellule senescenti. L’idea è che le cellule senescenti sopravvivono perché sovraesprimono proteine anti-apoptotiche come Bcl-2 e Bcl-XL. Quindi, inibendo queste proteine, dovremmo riuscire a eliminarle.
Abbiamo provato il navitoclax, un noto inibitore di Bcl-2/Bcl-XL/Bcl-w. Nei test a breve termine, sembrava funzionare, mostrando una certa tossicità selettiva per le cellule TIS. Ma nei trattamenti a lungo termine, i risultati erano ambigui, a volte funzionava, a volte no. Abbiamo anche provato a usarlo in un modello animale (topi con tumore al seno triplo negativo, molto aggressivo). La doxorubicina liposomiale (DOXIL) da sola induceva una risposta completa, facendo sparire il tumore per 40-60 giorni (rispecchiando il “risveglio” visto in vitro). Ma aggiungere navitoclax non migliorava per nulla la sopravvivenza degli animali.
Abbiamo testato altri inibitori: venetoclax (specifico per Bcl-2) non era selettivo per le TIS; ABT-737 (Bcl-2/Bcl-XL) e A-1331852 (Bcl-XL) mostravano una certa selettività, suggerendo che forse Bcl-XL fosse più importante di Bcl-2. Ma andando a misurare i livelli di queste proteine, non abbiamo trovato una correlazione chiara con l’efficacia degli inibitori. Anzi, spesso queste proteine erano già alte nelle cellule tumorali di partenza! Altri presunti senolitici (dasatinib, fisetina, quercetina, piperlongumina) si sono rivelati inefficaci contro le nostre cellule TIS. Insomma, la strada dei senolitici, almeno per come li conosciamo ora, sembrava un vicolo cieco per questo tipo di senescenza.
Dentro la Cellula Senescente: Cosa Cambia Davvero? (RNA-Seq)
Per capire cosa rendesse queste cellule TIS così diverse e resistenti, siamo andati a vedere quali geni venivano “accesi” o “spenti” (analisi del trascrittoma, o RNA-seq). Abbiamo confrontato le cellule di controllo (CTR), quelle senescenti (TIS) e quelle ripopolate (REPOP) per tutte e quattro le linee cellulari.
I risultati sono stati netti:
- Le cellule TIS avevano un profilo di espressione genica radicalmente diverso sia dalle CTR che dalle REPOP.
- Le cellule REPOP, invece, erano quasi identiche alle cellule CTR originali, confermando che il cambiamento era transitorio.
- Abbiamo identificato un set di 929 geni la cui espressione cambiava significativamente in TIS rispetto a CTR in tutte e quattro le linee cellulari. Sorprendentemente, la stragrande maggioranza (896) erano sovraespressi (accesi di più) e solo 33 sottoespressi (spenti).
- Nel passaggio da TIS a REPOP, avveniva il contrario: 706 geni venivano sottoespressi e solo 16 sovraespressi. Era come se la cellula “resettasse” il suo programma genetico.
- Abbiamo isolato 316 geni che erano sovraespressi esclusivamente nello stato TIS e tornavano normali in REPOP: una possibile “firma” molecolare della TIS nel cancro al seno.
Analizzando le funzioni di questi geni, abbiamo visto che le cellule TIS:
- Non proliferavano (come previsto, i geni legati alla divisione cellulare erano spenti).
- Mostravano un’attivazione delle vie di segnalazione legate a KRAS (un noto oncogene, anche se di solito non è mutato nel cancro al seno).
- Avevano una ridotta capacità di riparare il DNA.
- Presentavano alterazioni significative in tutte le vie legate alla risposta immunitaria (come Interferone, IL6-JAK-STAT3, Infiammazione). Questo suggerisce che le cellule TIS potrebbero manipolare il sistema immunitario per sfuggirgli.
Abbiamo fatto un confronto anche con cellule sane (fibroblasti di prepuzio umano, HFF) indotte in senescenza. Anche loro mostravano alcuni cambiamenti simili (es. KRAS), ma la risposta era meno marcata e alcune vie chiave (come quella del TNFα via NFκB) andavano in direzione opposta rispetto alle cellule tumorali.
Ma la cosa più frustrante è stata cercare una spiegazione alla resistenza ai farmaci nei geni. Abbiamo controllato l’espressione di decine di geni noti per causare resistenza:
- Pompe di efflusso (come ABCB1, ABCG2): alcune erano sovraespresse in TIS (ABCB1 e ABCG2 in tutte le linee), ma la loro specificità non spiegava perché alcuni farmaci funzionassero e altri no. Abbiamo provato a inibire ABCB1, ma non ha cambiato la sensibilità alla doxorubicina.
- Geni di riparazione del DNA: spesso erano sottoespressi, non sovraespressi.
- Target dei farmaci: l’espressione dei geni bersaglio dei farmaci testati non correlava quasi mai con la resistenza o la sensibilità osservata (es. TOP2A per gli inibitori della topoisomerasi, CDK per gli inibitori CDK, FLT3 per inibitori FLT3…).
- Geni che inattivano i farmaci o contrastano i loro effetti (es. RRM1/2, SAMHD1, DCK): nessun pattern chiaro.
In pratica, i meccanismi classici di farmaco-resistenza non sembravano essere i protagonisti principali in questo scenario. La resistenza sembrava una proprietà intrinseca dello stato TIS.
Uno Sguardo più da Vicino: L’Analisi a Singola Cellula (scRNA-Seq)
Per avere un quadro ancora più dettagliato, abbiamo usato l’RNA-seq a singola cellula (scRNA-seq) su due linee cellulari (MCF7 e T47D). Questo ci permette di vedere cosa succede in ogni singola cellula, invece che fare una media dell’intera popolazione.
I risultati hanno confermato e rafforzato quanto visto con l’analisi “bulk”:
- Le cellule TIS formavano un gruppo (cluster) completamente separato dalle cellule CTR e REPOP, evidenziando uno stato trascrizionale unico.
- Le cellule REPOP si raggruppavano molto vicino alle CTR, mostrando il ritorno allo stato originale.
- Abbiamo confermato l’alta espressione del marcatore di senescenza CDKN1A (p21) e la bassa espressione di LMNB1, TOP2A e MKI67 (marcatori di proliferazione) nelle cellule TIS.
- Interessante, abbiamo notato una piccola sottopopolazione di cellule TIS che sembrava co-esprimere marcatori sia di arresto che di potenziale riattivazione (“escapers”), forse il punto di partenza per la ripopolazione.
- L’analisi del ciclo cellulare ha mostrato che la stragrande maggioranza delle cellule TIS era bloccata in fase G1 (79.7% in MCF7, 66% in T47D), mentre CTR e REPOP avevano una distribuzione più equilibrata tra G1, S e G2/M.
- Le analisi delle vie metaboliche e di segnalazione hanno confermato la soppressione delle vie di riparazione del DNA e del ciclo cellulare, e l’attivazione di vie legate all’adesione e alla segnalazione estrogenica (nelle cellule che esprimono il recettore).
Abbiamo anche usato un’analisi chiamata “traiettoria” per mappare il percorso trascrizionale da CTR a TIS e da TIS a REPOP. Questo ha visualizzato chiaramente la transizione verso uno stato distinto (TIS) e il successivo ritorno verso lo stato iniziale (REPOP), sottolineando la reversibilità del processo. Anche rieseguendo l’analisi escludendo i geni del ciclo cellulare (per evitare artefatti), le traiettorie rimanevano le stesse.
Ancora una volta, guardando i geni di resistenza noti a livello di singola cellula, non abbiamo trovato un meccanismo comune che spiegasse la resistenza osservata. Ad esempio, TOP2A era basso in TIS MCF7 (potrebbe spiegare la resistenza agli inibitori topo II) ma alto in TIS T47D (che erano comunque resistenti). Un vero rompicapo!
Sulla Superficie della Battaglia: Il Proteoma di Superficie
Se i geni non ci davano la risposta completa, forse le proteine sì? In particolare, quelle sulla superficie della cellula o secrete all’esterno, che sono spesso coinvolte nella comunicazione con l’ambiente e nell’interazione con farmaci o sistema immunitario. Abbiamo usato una tecnica speciale (biotinilazione di superficie) per isolare e identificare queste proteine (analisi del proteoma di superficie) nelle cellule MCF7 (CTR, TIS, REPOP).
Anche qui, il profilo delle cellule TIS era nettamente diverso: 420 proteine erano espresse differentemente rispetto a CTR, 370 rispetto a REPOP, mentre tra CTR e REPOP le differenze erano solo 92.
Cosa abbiamo trovato di interessante sulla superficie o secreto dalle cellule TIS?
- Un sacco di proteine legate allo spliceosoma (il macchinario che processa l’RNA). Questo è stato inaspettato! Anche se si sa che lo splicing è alterato in senescenza, trovarne i componenti fuori dalla cellula è una novità e potrebbe indicare un nuovo meccanismo di comunicazione o resistenza. Ben 85 di queste proteine erano state trovate in vescicole extracellulari secrete da cellule tumorali senescenti in altri studi.
- Conferma dell’attivazione della via di KRAS. Addirittura, abbiamo trovato la proteina KRAS stessa arricchita nelle frazioni di membrana/secrete delle cellule TIS, nonostante l’mRNA non fosse aumentato e il gene non fosse mutato. Questo rafforza l’idea di un’attivazione “non canonica” di questa via, che potrebbe essere cruciale per l’escape dalla senescenza. Abbiamo provato un inibitore pan-KRAS, ma non ha mostrato tossicità selettiva, suggerendo che forse non è KRAS stesso il motore, ma qualche componente a valle della sua via.
- Molte proteine associate al SASP (Senescence-Associated Secretory Phenotype), il cocktail di molecole (citochine, chemochine, fattori di crescita, proteasi) che le cellule senescenti secernono per comunicare con l’ambiente. Tra queste, fattori già noti (come HMGB1, GDF15) e altri meno conosciuti.
- Un gruppo di proteine legate alla biogenesi dei ribosomi, confermando studi precedenti che collegano difetti nella produzione di ribosomi alla senescenza.
Abbiamo anche identificato 52 proteine mai associate prima alla senescenza, che potrebbero essere nuovi marcatori o avere ruoli funzionali nello stato TIS.
Tentativi Falliti e Nuove Speranze
Armati di questi dati, abbiamo provato a colpire alcune delle proteine più sovraespresse specificamente in TIS (sia a livello di mRNA che, in alcuni casi, di proteina) per le quali esistevano inibitori: KRT6A (cheratina 6A, inibita da simvastatina), PSMA8 (subunità proteasoma, inibita da MG132), PDE1A (fosfodiesterasi, inibita da timochinone), GSDMC (gasdermina C, attivata da DM-αKG/Actinomicina D), ACSM2A (acil-CoA sintetasi, inibita da acido 4-metilsalicilico). Purtroppo, nessuno di questi tentativi ha funzionato: gli inibitori o non erano tossici, o uccidevano sia le cellule TIS che le CTR senza selettività. Questo rafforza l’idea che la resistenza della TIS sia un fenomeno complesso e non legato a un singolo bersaglio facilmente attaccabile.
Implicazioni e Prospettive Future: Un Nemico Nascosto
Quindi, cosa significa tutto questo? La senescenza indotta da terapia, almeno nel cancro al seno che abbiamo studiato, non sembra essere quella “soluzione” definitiva che speravamo. Anzi, si configura come un astuto meccanismo di resistenza transitoria. Le cellule entrano in uno stato di “animazione sospesa” che le protegge da un’ampia gamma di farmaci e, probabilmente, anche dal sistema immunitario, per poi risvegliarsi e riprendere la crescita quando le condizioni migliorano.
Questo ha implicazioni enormi:
1. La TIS non è un endpoint terapeutico desiderabile: Indurre senescenza potrebbe non bastare, anzi, potrebbe selezionare cellule pronte a tornare più aggressive.
2. La resistenza TIS è complessa: Non dipende dai soliti meccanismi noti, ma sembra una proprietà emergente dello stato senescente stesso, forse legata a cambiamenti epigenetici profondi o modifiche post-traduzionali delle proteine che ancora non comprendiamo appieno.
3. L’evasione immunitaria è cruciale: Le cellule TIS sembrano specializzarsi nel manipolare il sistema immunitario. Esprimono fattori (come galectina 9, CXCL12, TREM1) che possono silenziare le cellule T killer o creare uno scudo protettivo. Anche le cellule REPOP mantengono una “memoria” immunitaria alterata (geni come IFITs, OAS), che potrebbe aiutarle in future ricadute.
4. Nuovi bersagli? Sebbene i senolitici classici e gli inibitori mirati ai geni sovraespressi abbiano fallito, forse potremmo colpire le vulnerabilità specifiche della TIS:
* I meccanismi di evasione immunitaria (es. anticorpi contro LGALS9 o TREM1?).
* Le proteine uniche di superficie identificate dalla proteomica (potrebbero essere target per immunoterapie come CAR-T o anticorpi coniugati a farmaci?).
* La via di KRAS attivata in modo non canonico (se capiamo come viene attivata, potremmo bloccarla?).
* I meccanismi epigenetici o legati allo splicing che mantengono lo stato TIS.
Insomma, la battaglia contro il cancro si rivela ancora una volta più complessa. La senescenza indotta da terapia è un Giano bifronte: da un lato ferma la crescita, dall’altro nasconde una resistenza subdola e prepara il terreno per la ricaduta. Capire a fondo questo meccanismo, con le sue stranezze molecolari e le sue strategie di evasione, è fondamentale per sviluppare terapie future che possano davvero sradicare il tumore, senza lasciare cellule “dormienti” pronte a risvegliarsi. La ricerca continua!
Fonte: Springer