Rabdomiosarcoma Pleomorfo: Il Camaleonte dei Tumori Rari negli Adulti
Ciao a tutti! Oggi voglio parlarvi di qualcosa di piuttosto insolito nel mondo dell’oncologia, qualcosa che spesso si nasconde sotto mentite spoglie: il rabdomiosarcoma pleomorfo (PRMS) negli adulti. Magari avete sentito parlare di rabdomiosarcoma nei bambini – lì è il tipo più comune di sarcoma dei tessuti molli. Ma negli adulti? È tutta un’altra storia, molto più rara e, purtroppo, spesso più aggressiva.
Pensate che i sarcomi dei tessuti molli rappresentano meno dell’1% di tutti i tumori negli adulti, e il rabdomiosarcoma è solo una piccola fetta (circa il 3%) di quella già piccola torta. Ma quando colpisce, può farlo ovunque, anche dove non ci aspetteremmo di trovare tessuto muscolare scheletrico. Strano, vero?
Un Tumore dai Mille Volti
Il rabdomiosarcoma non è tutto uguale. Esistono principalmente tre sottotipi: embrionale, alveolare e, appunto, pleomorfo. Mentre la forma embrionale è più comune nei piccoli e ha una prognosi generalmente migliore, e quella alveolare può colpire a tutte le età (spesso legata a specifiche traslocazioni genetiche come quelle che coinvolgono i geni PAX3 o PAX7 e FKHR), la variante pleomorfa è tipica degli adulti sopra i 45-50 anni. Ed è proprio questa forma ad avere la reputazione peggiore, caratterizzata da una tendenza a dare metastasi precocemente.
L’origine esatta di questo tumore è ancora un po’ un mistero. Anche se imita il tessuto muscolare scheletrico in via di sviluppo, ricerche recenti suggeriscono che potrebbe nascere da cellule non muscolari che prendono una strada sbagliata durante il loro sviluppo. Le sedi più comuni sono testa e collo, tratto genitourinario ed estremità. Trovarlo nel tratto gastrointestinale come tumore primario? Estremamente raro. Di solito, se c’è, è una metastasi da altrove.
Un Caso Clinico Emblematico: Quando la Diagnosi è un Puzzle
Voglio raccontarvi di un caso che illustra perfettamente le sfide legate a questo tumore. Un uomo iraniano di 67 anni si presenta in ospedale. I suoi sintomi? Debolezza e dolore nella parte alta dell’addome (epigastrio) da circa tre mesi. Gli esami del sangue rivelano subito un problema serio: anemia grave, con un’emoglobina bassissima (3 g/dL!).
Indagini più approfondite, come ecografia e TAC con contrasto, mostrano una massa polipoide di dimensioni considerevoli (circa 8×7 cm) tra lo stomaco (antro) e il duodeno, oltre a linfonodi ingrossati nel mediastino. La gastroscopia conferma la presenza di questa grossa massa nel duodeno. Viene fatta una biopsia, un piccolo prelievo di tessuto. L’analisi iniziale al microscopio suggerisce un tumore stromale gastrointestinale (GIST) epiteliode poco differenziato. Il GIST è un tipo di sarcoma più comune nel tratto GI, quindi è un sospetto logico. Ma qualcosa non torna del tutto, serve indagare meglio.

L’uomo viene quindi operato: gli asportano completamente lo stomaco e collegano l’intestino (gastrodigiunostomia). L’intervento va bene. Ora si può esaminare l’intera massa tumorale, non solo un piccolo frammento. Ed è qui che la vera natura del tumore viene a galla.
La Chiave è nell’Immunoistochimica
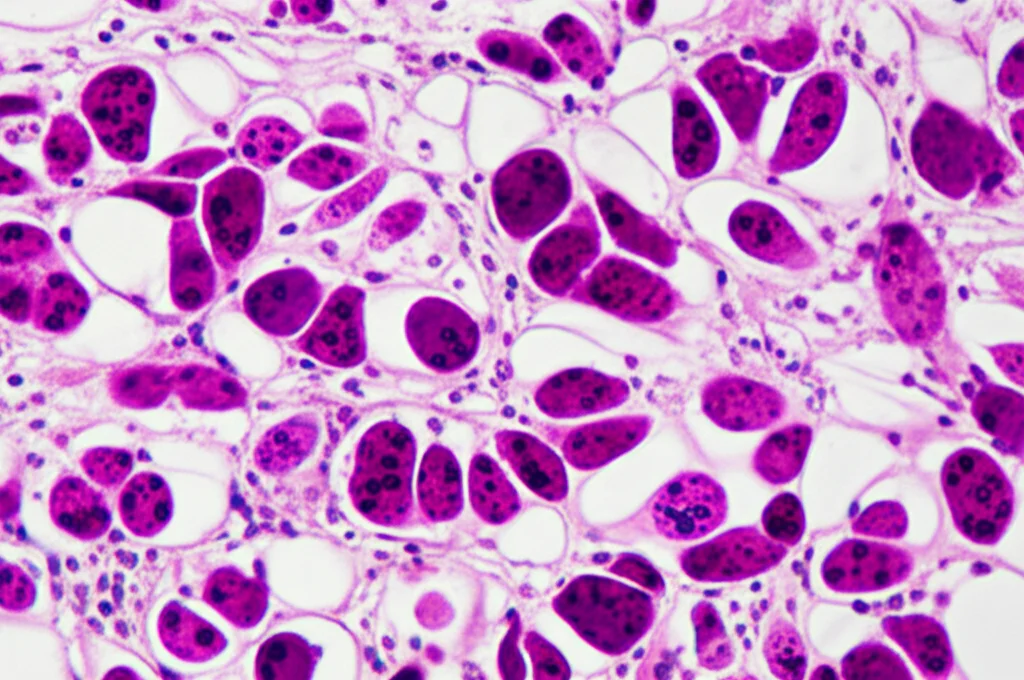
L’esame al microscopio del pezzo operatorio mostra un quadro diverso dalla biopsia iniziale. Si vedono cellule molto variegate (pleomorfe), alcune rotonde, altre allungate (fusiformi), con un citoplasma rosa (eosinofilo), decisamente bizzarre, che mostrano segni di differenziazione verso il muscolo scheletrico. Il tumore si estende fino alla sottomucosa, ma per fortuna non sembra aver invaso i vasi sanguigni o linfatici.
A questo punto, i patologi si trovano di fronte a diverse possibilità: linfoma, melanoma mucoso, carcinoma indifferenziato, GIST epiteliode, altri sarcomi… Come distinguere? Qui entra in gioco una tecnica fondamentale: l’immunoistochimica (IHC). Si usano anticorpi specifici per “colorare” determinate proteine presenti (o assenti) nelle cellule tumorali, creando un profilo unico per ogni tipo di tumore.
Nel nostro caso, i risultati dell’IHC sono stati decisivi:
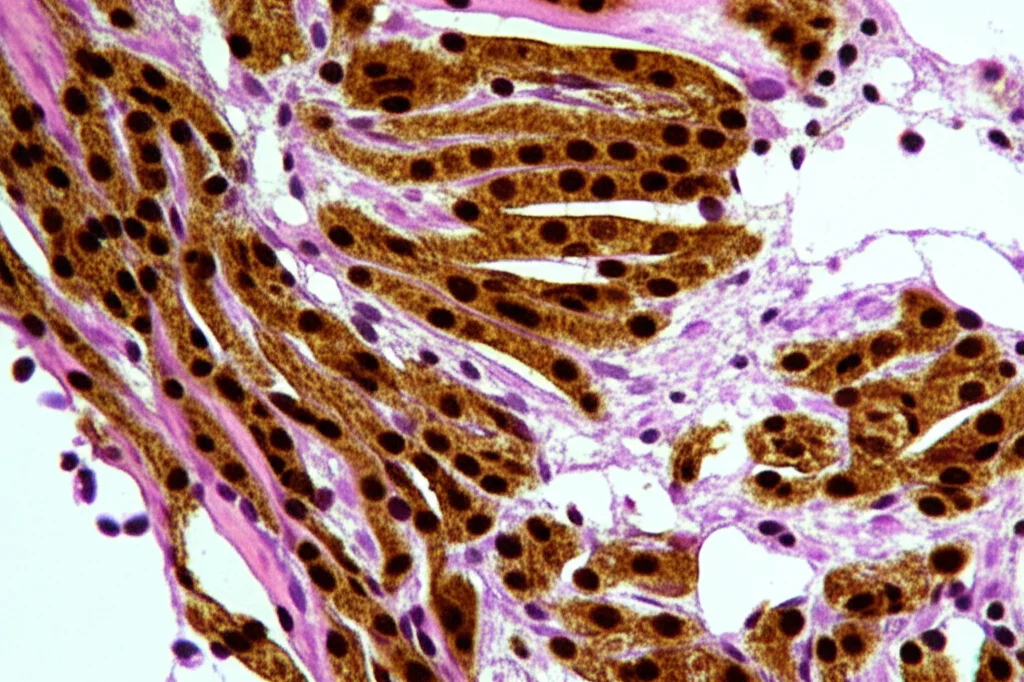
- Positività diffusa per: Desmina, Vimentina (marcatori muscolari e mesenchimali)
- Positività per: Miogenina (marcatore specifico della differenziazione muscolare scheletrica)
- Positività sparsa per: CD45 (presente in alcune cellule infiammatorie, ma la sua presenza sparsa non indica un linfoma)
- Negatività per: CD34, c-KIT (CD117), DOG1 (marcatori tipici dei GIST), S100, Melan-A (marcatori per melanoma e tumori neurali), Citocheratina (marcatore per carcinomi)
Questo profilo immunofenotipico esclude le altre ipotesi e punta dritto a una diagnosi: Rabdomiosarcoma Pleomorfo primario del duodeno. È stato anche eseguito un test molecolare per le fusioni geniche PAX3/7-FKHR, risultato negativo, il che è coerente con la diagnosi di PRMS (quelle fusioni sono tipiche della variante alveolare).
Gestione e Prospettive Future
Il trattamento principale per il PRMS, specialmente nel tratto gastrointestinale, è la chirurgia radicale, con l’obiettivo di rimuovere completamente il tumore. Tuttavia, data l’aggressività del PRMS e la frequente presenza di micro-metastasi già al momento della diagnosi, spesso si ricorre a terapie aggiuntive (adiuvanti).
Si stanno valutando regimi di chemioterapia (spesso con farmaci come vincristina, dactinomicina e ciclofosfamide) e la radioterapia può essere usata se i margini chirurgici sono a rischio o se la resezione completa non è stata possibile.
Un campo in grande sviluppo è la profilazione molecolare del tumore. Identificare specifiche alterazioni genetiche può aiutare a scegliere terapie mirate, personalizzando il trattamento. Questo è cruciale data l’eterogeneità del PRMS e la mancanza, ad oggi, di protocolli standardizzati universalmente accettati.
Nel caso del nostro paziente, dopo 12 mesi dall’intervento, fortunatamente non c’erano segni di recidiva locale o metastasi a distanza. Una buona notizia, ma sappiamo che la prognosi a lungo termine per il PRMS gastrointestinale rimane generalmente sfavorevole. La rarità della malattia, la diagnosi spesso tardiva e l’alto potenziale metastatico sono ostacoli importanti.
Cosa Impariamo da Questo Caso?
Questo caso ci insegna molto. Innanzitutto, che bisogna pensare al rabdomiosarcoma anche in sedi insolite come il duodeno e negli adulti, specialmente di fronte a neoplasie poco differenziate. In secondo luogo, sottolinea il ruolo cruciale dell’immunoistochimica per arrivare a una diagnosi corretta, distinguendo il PRMS da altri tumori con aspetto simile, come i GIST.
La strada per migliorare la gestione di questi tumori rari è ancora lunga. Servono più ricerca, studi multicentrici per raccogliere più dati, una maggiore consapevolezza tra i medici e lo sviluppo di protocolli di trattamento più efficaci e standardizzati, magari sfruttando le nuove conoscenze di genomica e le terapie mirate. Solo così potremo sperare di migliorare la prognosi per i pazienti che si trovano ad affrontare questo “camaleonte” dei tumori.
Fonte: Springer