
QDπ: Il Tesoro di Dati che Rivoluziona la Scoperta di Farmaci con l’IA
Avete mai pensato a come nasce un nuovo farmaco? È un percorso lungo, complesso e costoso. Ma oggi, grazie all’intelligenza artificiale (IA) e ai modelli di machine learning (ML), stiamo assistendo a una vera e propria rivoluzione. Immaginate di poter simulare il comportamento di milioni di molecole potenzialmente terapeutiche in modo rapido e accurato. Fantascienza? Non proprio! Ma c’è un “ma”: per funzionare bene, questi modelli di IA, chiamati Machine Learning Potentials (MLP), hanno bisogno di una quantità enorme di dati di altissima qualità. Dati che descrivano come si comportano le molecole, le loro energie, le forze tra gli atomi. E non dati qualsiasi, ma dati diversificati e accurati.
La Sfida: Dati, Dati Ovunque, Ma Quali Scegliere?
Il problema è che creare questi dataset è un lavoro immane. Fino ad oggi, esistevano diversi dataset, ma spesso con dei limiti:
- Alcuni erano troppo “stretti”, coprivano cioè una varietà limitata di molecole.
- Altri usavano metodi di calcolo quantistico (QM) non abbastanza precisi, introducendo errori che poi l’IA imparava.
- Altri ancora, pur essendo enormi, contenevano molte informazioni ridondanti, rendendo l’addestramento dell’IA inefficiente e costoso.
Pensateci: calcolare l’energia e le forze di milioni di conformazioni molecolari con metodi quantistici accurati richiede una potenza di calcolo spaventosa! Come fare, quindi, a creare un dataset che sia allo stesso tempo ampio, accurato e senza sprechi?
La Nostra Soluzione: Nasce il QDπdataset!
Ed è qui che entriamo in gioco noi, con il QDπdataset (Quantum Deep Potential Interaction dataset). Cosa abbiamo fatto? Abbiamo deciso di creare un nuovo dataset “intelligente”, pensato apposta per addestrare MLP universali nel campo della scoperta di farmaci.
Non siamo partiti da zero. Abbiamo preso il meglio da diversi dataset già esistenti (come SPICE, ANI, GEOM, FreeSolv, RE, COMP6) e li abbiamo “fusi” insieme. Ma non in modo casuale! Abbiamo usato una strategia chiamata apprendimento attivo (active learning), in particolare una tecnica detta “query-by-committee”.
Immaginate un comitato di esperti (i nostri modelli MLP preliminari) che esamina ogni singola struttura molecolare proveniente dai dataset originali. Se gli esperti sono tutti d’accordo su come si comporta quella molecola, significa che probabilmente non aggiunge molte informazioni nuove rispetto a quelle che già abbiamo. Quindi, la scartiamo! Se invece gli esperti “litigano”, cioè danno previsioni diverse, allora quella struttura è interessante, contiene informazioni preziose e la includiamo nel nostro QDπdataset, ricalcolandone le proprietà con un metodo quantistico molto accurato: il ωB97M-D3(BJ)/def2-TZVPPD.
Questo metodo è considerato uno dei migliori compromessi tra accuratezza e costo computazionale, specialmente per le interazioni non covalenti, cruciali nel mondo dei farmaci e delle biomolecole. Abbiamo verificato che usare metodi meno precisi, come quello usato per i dataset ANI originali, introduce differenze significative (anche più di 2-3 kcal/mol/Å nelle forze!) che possono compromettere l’affidabilità dell’IA.
Cosa C’è Dentro QDπ? Un Mix Esplosivo di Diversità Chimica
Grazie a questa strategia “smart”, siamo riusciti a condensare l’informazione chimica rilevante in “soli” 1.6 milioni di strutture molecolari. Sembrano tante, ma sono molte meno di quelle contenute nei dataset originali presi singolarmente, pur mantenendo (e anzi, aumentando!) la diversità chimica.
Il QDπdataset copre ben 13 elementi chimici (H, C, N, O, F, P, S, Cl, Br, I, e anche Li, Na, K, importanti per ioni e interazioni), fondamentali per le molecole organiche, i farmaci e i frammenti di biopolimeri (come amminoacidi e peptidi).
Cosa contiene esattamente?
- Molecole neutre e cariche: Un punto debole di molti MLP era la gestione degli stati di carica e protonazione. Noi abbiamo incluso dati specifici per migliorare questo aspetto.
- Conformazioni diverse: Non solo geometrie ottimizzate (stabili), ma anche conformazioni accessibili termicamente, ottenute tramite simulazioni di dinamica molecolare (MD). Questo è fondamentale perché le molecole non sono statiche, si muovono e cambiano forma!
- Interazioni intermolecolari: Dati cruciali per capire come un farmaco interagisce con il suo bersaglio biologico.
- Tautomeri ed energie di protonazione relative: Informazioni importantissime per la chimica farmaceutica.
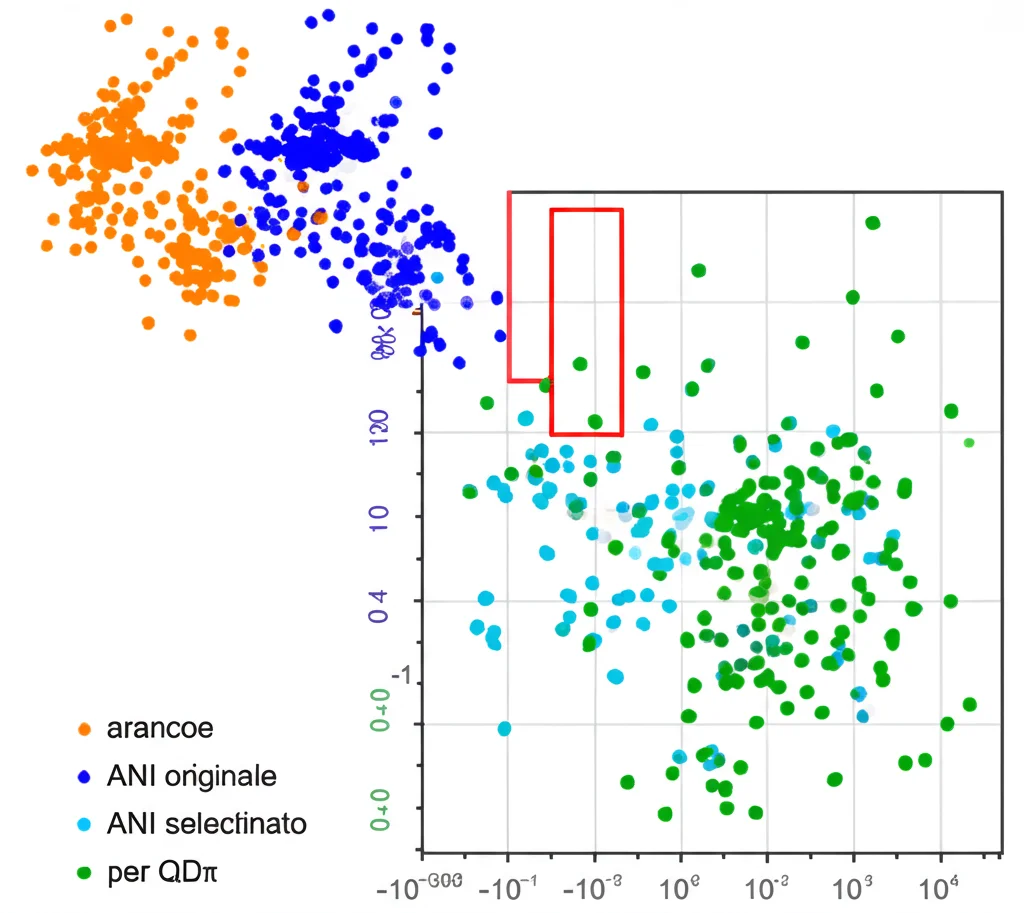
Abbiamo analizzato la diversità chimica del nostro dataset usando una tecnica chiamata t-SNE. È come creare una mappa 2D dello spazio chimico coperto dai dati. Quello che abbiamo visto è che QDπ copre aree che né SPICE né ANI da soli riuscivano a coprire completamente. L’apprendimento attivo ha fatto un ottimo lavoro nel selezionare solo le informazioni “nuove” da ANI, evitando le ridondanze con SPICE.
Perché QDπ è Importante per Voi (e per la Scienza)
Il nostro obiettivo è chiaro: fornire alla comunità scientifica una risorsa preziosa per sviluppare la prossima generazione di MLP universali per la scoperta di farmaci. Con QDπ, speriamo di:
- Accelerare lo screening virtuale: Permettere di valutare più molecole candidate in meno tempo e con maggiore accuratezza.
- Migliorare la predizione delle proprietà: Ottenere stime più affidabili di energie conformazionali, barriere energetiche, interazioni, ecc.
- Facilitare lo sviluppo di nuovi farmaci: Contribuire a superare le limitazioni dei metodi tradizionali.
Il bello è che il QDπdataset è liberamente accessibile! Lo abbiamo reso disponibile su Zenodo in formato HDF5, compatibile con software popolari come DeePMD-kit. Abbiamo anche fornito un semplice script Python per mostrarvi come caricarlo e iniziare a usarlo.
Guardando al Futuro
Siamo convinti che la densità e la diversità di informazioni chimiche contenute nel QDπdataset rappresentino un passo avanti significativo. È uno strumento potente che, speriamo, ispirerà e abiliterà la creazione di modelli IA ancora più sofisticati e accurati, portandoci sempre più vicini a progettare farmaci migliori in modo più efficiente. Il viaggio è appena iniziato!
Fonte: Springer





