Proteomica Low-Input: La Rivoluzione Accessibile della Trappola Ionica Lineare!
Ciao a tutti! Oggi voglio parlarvi di una cosa che mi appassiona tantissimo nel mio campo, la proteomica, e di come stiamo cercando di renderla più accessibile e potente, soprattutto quando abbiamo a che fare con quantità minuscole di materiale biologico. Immaginate di voler studiare come comunicano tra loro piccoli gruppi di cellule nel nostro corpo, ad esempio nel sistema immunitario. È una sfida enorme, perché queste cellule sono poche e le proteine che vogliamo analizzare sono presenti in quantità infinitesimali.
Il Problema: Piccoli Campioni, Grandi Sfide
La biologia dei sistemi cerca proprio di capire queste interazioni complesse. Pensate al sistema immunitario adattativo: è un balletto incredibile di diversi tipi di cellule specializzate che lavorano insieme. Per capire come funziona questo balletto, dobbiamo poter analizzare le proteine specifiche di queste piccole popolazioni cellulari. Negli ultimi anni, la proteomica e la spettrometria di massa (MS) hanno fatto passi da gigante, permettendoci di avvicinarci all’analisi di poche cellule (low-input proteomics) o addirittura di cellule singole.
Finora, per fare questo tipo di analisi super sensibili, ci si è affidati principalmente a strumenti molto sofisticati e costosi, quelli con un’altissima accuratezza di massa, come gli Orbitrap. Sono macchine fantastiche, non c’è dubbio. Dall’altra parte, ci sono i tripli quadrupoli: strumenti super veloci e sensibili per analisi mirate (targeted proteomics), ma con una specificità di massa inferiore e, soprattutto, limitati a cercare solo quello che già sappiamo di dover cercare (tramite tecniche come l’SRM – Selected Reaction Monitoring). In pratica, con un triplo quadrupolo non puoi fare una scoperta “a sorpresa”, devi dirgli tu cosa misurare, e spesso le informazioni su cosa misurare devi ottenerle prima con uno strumento ad alta risoluzione.
La Soluzione: La Magia della Trappola Ionica Lineare (LIT)
E se ci fosse una via di mezzo? Uno strumento versatile, più economico, ma comunque potente? Ecco che entra in gioco la trappola ionica lineare (LIT). Le LIT sono analizzatori di massa nominale (cioè con una risoluzione di massa inferiore rispetto agli Orbitrap, simile ai tripli quadrupoli), ma hanno un asso nella manica: sono incredibilmente versatili. A differenza dei tripli quadrupoli, che misurano solo specifiche transizioni ione precursore/frammento, le LIT possono acquisire spettri MS/MS completi molto rapidamente. Questo le rende adatte sia per la proteomica mirata (usando una tecnica chiamata PRM – Parallel Reaction Monitoring) sia per quella globale, “esplorativa” (usando DDA o DIA – Data-Independent Acquisition).
Noi abbiamo pensato: e se usassimo uno strumento ibrido, un quadrupolo-LIT (Q-LIT), come soluzione “tutto-in-uno”? Uno strumento capace di fare entrambe le cose, scoperta e quantificazione mirata, senza bisogno dell’altissima accuratezza di massa e, quindi, con costi potenzialmente inferiori? Le LIT sono note per essere molto sensibili, perfette per i campioni “low-input” (diciamo sotto i 100 nanogrammi per iniezione), e in alcuni casi, specialmente sotto i 10 ng, possono addirittura superare gli analizzatori ad alta risoluzione! Inoltre, hanno requisiti di vuoto meno stringenti, il che significa strumenti potenzialmente più semplici, compatti ed economici.

Come Funziona il Nostro Approccio? Un Flusso di Lavoro Rapido
Abbiamo quindi messo a punto un flusso di lavoro che sfrutta proprio un Q-LIT (nello specifico, uno strumento Stellar™ della Thermo Scientific™). L’idea è usare lo stesso strumento sia per generare una “libreria” di peptidi presenti nel campione (usando la DIA, in particolare una variante chiamata GPF-DIA, Gas-Phase Fractionation DIA), sia per poi effettuare le misure quantitative mirate (PRM) sui bersagli che ci interessano di più.
La vera chicca è che abbiamo sviluppato un software open-source (integrato nel nostro tool EncyclopeDIA) che automatizza gran parte del processo. Questo software prende i dati DIA, identifica i peptidi, e crea automaticamente un “programma” ottimizzato per l’analisi PRM sullo stesso strumento. Chiamiamo questa libreria intermedia una “translation library”, perché “traduce” le informazioni dalla scoperta globale (DIA) al formato necessario per l’analisi mirata (PRM), tenendo conto delle caratteristiche specifiche dello strumento LIT (tempi di ritenzione, pattern di frammentazione a bassa risoluzione). Il bello è che tutto questo processo, dalla generazione della libreria all’ottenimento dell’analisi PRM pronta per essere eseguita, può essere fatto in una sola giornata lavorativa! Niente più bisogno di passare da uno strumento ad alta risoluzione a uno a bassa risoluzione, validando tutto da capo.
Mettiamolo alla Prova: Accuratezza e Sensibilità
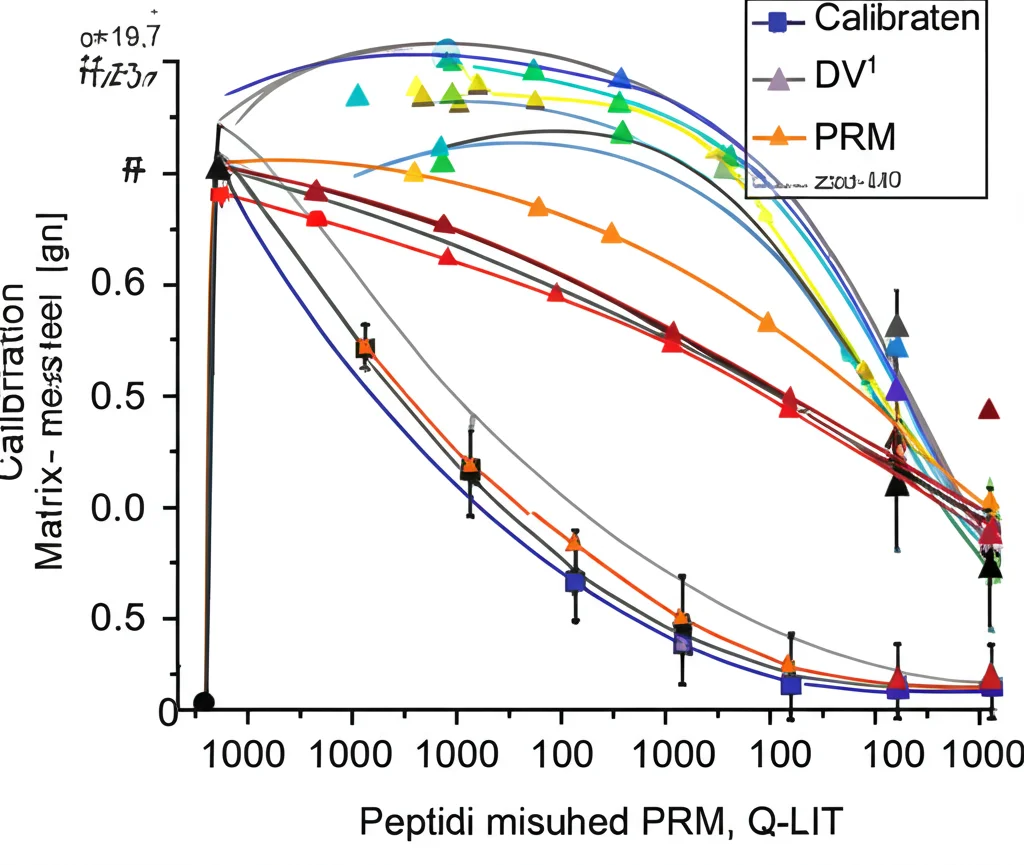
Ok, bello sulla carta, ma funziona davvero? Quantifica bene? Per verificarlo, abbiamo fatto degli esperimenti tosti. Abbiamo creato delle curve di calibrazione “matrix-matched”. Cosa significa? Invece di diluire semplicemente i peptidi che ci interessano in un buffer pulito (troppo facile!), li abbiamo diluiti in un background complesso, fatto dello stesso tipo di proteine del nostro campione, ma “marcate” chimicamente (con etichettatura dimetilica) in modo che non interferissero con la misura dei nostri target. Questo simula molto meglio una situazione reale.
Abbiamo testato tutto questo a diversi livelli di input: 100 ng, 10 ng e persino 1 ng di proteoma totale. I risultati? Fantastici! Con il Q-LIT, abbiamo ottenuto una quantificazione lineare e consistente su quasi tre ordini di grandezza di concentrazione. Anche con solo 1 ng di materiale (che è la quantità di proteine che ci si aspetta da circa 3-10 cellule!), siamo riusciti a quantificare proteine a bassa abbondanza, come fattori di trascrizione e citochine, con buona linearità. Ad esempio, un peptide della Granzima B (una proteina importante nelle cellule T citotossiche) è risultato quantificabile fino a livelli equivalenti a circa 43 picogrammi (0.043 ng) in un background di 1 ng!
Abbiamo anche confrontato le prestazioni del Q-LIT con quelle di un Q-Orbitrap (un Exploris 480, uno strumento ad alta risoluzione). Come ci aspettavamo da lavori precedenti, a bassissimi input (sotto i 10 ng), il Q-LIT ha identificato più proteine in modalità DIA. Nelle analisi PRM, il Q-LIT ha mostrato limiti di quantificazione (LoQ) mediamente migliori rispetto all’Orbitrap, specialmente ai livelli più bassi, e grazie alla sua velocità di scansione, ci ha permesso di monitorare più peptidi nello stesso tempo.
Un Esempio Concreto: Linfociti T sotto la Lente
Per vedere se questo approccio fosse utile in un contesto biologico reale, abbiamo studiato dei linfociti T umani. I linfociti T sono cellule chiave del sistema immunitario. Li abbiamo coltivati in laboratorio stimolandoli con due diverse citochine, l’Interleuchina-2 (IL-2) e l’Interleuchina-15 (IL-15). È noto che queste due citochine, pur essendo simili e usando parte dello stesso recettore, spingono i linfociti T verso destini leggermente diversi: IL-2 favorisce uno stato più “effettore”, mentre IL-15 uno stato più “di memoria”.
Abbiamo analizzato campioni da 1 ng di queste cellule T stimolate usando il nostro metodo Q-LIT PRM, focalizzandoci su proteine importanti per l’attivazione, la differenziazione e la segnalazione delle citochine. In parallelo, abbiamo analizzato le stesse popolazioni cellulari con la citometria a flusso, una tecnica standard per caratterizzare le cellule immunitarie basata su marcatori di superficie.
I risultati della proteomica mirata con Q-LIT si sono rivelati incredibilmente consistenti con i dati della citometria a flusso! Ad esempio, la citometria mostrava una percentuale maggiore di cellule CD8+ nel campione stimolato con IL-15 rispetto a quello con IL-2, e una percentuale minore di cellule CD4+. La nostra analisi PRM ha confermato questo: la proteina CD4 era significativamente meno abbondante nel campione IL-15. Anche se tecnicamente il peptide CD4 era sotto il limite di quantificazione a 1 ng (ma sopra il limite di rilevamento), la tendenza era chiara e confermata dalle misure a 10 ng. Abbiamo anche visto differenze nell’espressione di Granzima B (più alta con IL-2, coerente con più cellule effettrici) e di marcatori associati allo stato di memoria (come CD62L, che la citometria mostrava più alto con IL-15, e IL-2Rβ, che la proteomica mostrava più alto con IL-2). La precisione tecnica delle misure PRM a 1 ng era ottima, con la maggior parte dei peptidi misurati con un coefficiente di variazione inferiore al 20% tra repliche tecniche.

Perché Tutto Questo è Importante?
Questi risultati ci dicono che gli strumenti Q-LIT rappresentano una soluzione davvero valida e potente per espandere l’uso della spettrometria di massa in molti laboratori. Sono strumenti:
- Versatili: Capaci di fare sia scoperta (DIA) che quantificazione mirata (PRM).
- Sensibili: Ottimi per campioni low-input, anche a livello di poche cellule.
- Rapidi: Permettono di sviluppare saggi mirati in tempi brevi.
- Accessibili: Potenzialmente più economici da acquistare e mantenere rispetto agli strumenti ad altissima risoluzione.
Questo ultimo punto è cruciale. Rendere la proteomica avanzata più accessibile può avere un impatto enorme, specialmente in campi come l’immuno-oncologia, dove analizzare le popolazioni di cellule immunitarie a livello proteico può svelare biomarcatori fondamentali per guidare le decisioni cliniche.
Insomma, credo fermamente che queste “vecchie” ma rivitalizzate trappole ioniche lineari abbiano ancora molto da dire e possano davvero democratizzare la proteomica di precisione. È un momento entusiasmante per essere in questo campo!
Fonte: Springer