
MOF e Fononi: L’Intelligenza Artificiale Svela i Segreti Vibrazionali dei Materiali del Futuro!
Amici appassionati di scienza e scoperte, oggi voglio parlarvi di qualcosa che mi sta particolarmente a cuore e che, credetemi, sta per rivoluzionare il modo in cui progettiamo i materiali del futuro. Sto parlando dei Metal-Organic Frameworks (MOF) e di come, grazie a un pizzico di intelligenza artificiale, stiamo finalmente riuscendo a svelare alcuni dei loro segreti più intimi, legati alle loro vibrazioni atomiche, i cosiddetti fononi.
Cosa sono i MOF e perché ci piacciono tanto?
Immaginate dei mattoncini LEGO® su scala nanometrica. I MOF sono proprio così: strutture cristalline incredibilmente porose, formate da ioni metallici (i “nodi”) collegati da molecole organiche (i “linker”). Questa loro natura modulare e la loro elevatissima porosità li rendono candidati ideali per un sacco di applicazioni fichissime: dalla cattura dell’anidride carbonica (per combattere il cambiamento climatico, mica poco!) alla raccolta di acqua dall’atmosfera (pensate alle zone aride!), passando per la catalisi e i biosensori. Insomma, dei veri e propri tuttofare della chimica!
Ma c’è un “ma”. Per sfruttare al meglio questi materiali, dobbiamo capire a fondo le loro proprietà fisiche, come la stabilità meccanica, l’espansione termica, la conduzione del calore e persino la superconduttività. E qui entrano in gioco i fononi. I fononi, in parole povere, descrivono le vibrazioni collettive degli atomi in un cristallo. Sono loro che influenzano pesantemente tutte queste belle proprietà. Il problema? Studiarli nei MOF è un vero e proprio rompicapo.
Il problema dei fononi: un osso duro per i metodi tradizionali
Tradizionalmente, per studiare queste cose, ci si affida a metodi computazionali potentissimi come la Density Functional Theory (DFT). La DFT è fantastica, una vera roccia per noi scienziati dei materiali. Però, quando hai a che fare con i MOF, che possono avere centinaia o addirittura migliaia di atomi nella loro cella unitaria (l’unità base che si ripete per formare il cristallo), la DFT diventa un incubo computazionale. Immaginate di dover fare calcoli su supercelle enormi: ci vorrebbero secoli e risorse infinite, soprattutto se vuoi fare uno screening ad alto rendimento, cioè analizzare tantissimi MOF diversi per trovare quello giusto per una specifica applicazione.
Certo, esistono alternative più “leggere”:
- Metodi quanto-meccanici semi-empirici come il Density Functional Tight Binding (DFTB): sono più veloci, ma la loro parametrizzazione (cioè l’adattamento dei parametri del modello) e la mancanza di parametri per alcuni atomi metallici ne limitano l’uso, specialmente per i fononi nei MOF. Una variante, GFN1-xTB, è più versatile ma ha ancora i suoi limiti per le proprietà vibrazionali.
- Campi di forza tradizionali (Force Fields) come UFF o CHARMM: super veloci e scalabili, ma spesso peccano in accuratezza quando si tratta di prevedere proprietà dinamiche come i fononi. Anche versioni specifiche per i MOF, come UFF4MOF o MOF-FF, pur essendo utili, mostrano i loro limiti. Addirittura, un modello focalizzato sulle vibrazioni come VMOF, pur riproducendo bene alcune cose, sottostimava di brutto il modulo di comprimibilità (una misura di quanto un materiale si “schiaccia” sotto pressione) rispetto alla DFT.
Il problema di fondo con i campi di forza classici è l’immensa varietà chimica dei MOF: ottimizzare i parametri per tutte le combinazioni possibili è una sfida titanica.

Entra in gioco l’Intelligenza Artificiale: i Potenziali di Machine Learning (MLP)
Ed è qui che l’intelligenza artificiale ci viene in soccorso! Negli ultimi anni, sono emersi i potenziali di machine learning (MLP), modelli che imparano le relazioni tra la struttura atomica e l’energia del sistema direttamente dai dati DFT. Alcuni di questi, come quelli “on-the-fly” (cioè che si addestrano continuamente), hanno mostrato buona accuratezza per le proprietà vibrazionali dei MOF. Però, c’è un inghippo: sono spesso limitati allo specifico MOF per cui sono stati addestrati e richiedono una continua rigenerazione e riaddestramento con nuovi dati DFT. Non proprio l’ideale per lo screening ad alto rendimento, vero?
Ecco perché c’era un disperato bisogno di un modello “pronto all’uso” e trasferibile. E qui entra in scena un protagonista interessante: il modello MACE (specifically MACE-MP-0). Questo modello, basato su una rete tensoriale a grafo con passaggio di messaggi equivariante (lo so, suona complicato, ma fidatevi, è roba forte!), è stato addestrato su un vasto dataset di cristalli inorganici. Sorprendentemente, ha dimostrato una buona capacità di predire l’energia anche per i MOF, suggerendo che potesse essere un’ottima base di partenza per studiare i fononi.
MACE-MP-MOF0: il nostro asso nella manica!
Partendo da questa solida base (una versione leggermente modificata chiamata MACE-MP-0b), nel nostro lavoro abbiamo sviluppato MACE-MP-MOF0. Si tratta di un modello MACE “raffinato” (fine-tuned, in gergo tecnico) specificamente per i MOF. Come abbiamo fatto? Abbiamo creato un dataset di alta qualità composto da 127 MOF, scelti per essere rappresentativi e diversi tra loro. Questi MOF coprono un’ampia gamma di 24 elementi chimici diversi, presenti sia nei nodi inorganici che nei linker organici, e tutte e 7 le simmetrie cristalline.
Il risultato? MACE-MP-MOF0 è una bomba! Non solo migliora significativamente l’accuratezza della densità di stati fononici (una sorta di “impronta digitale” delle vibrazioni del materiale) rispetto al modello di partenza, ma corregge anche i fastidiosissimi “modi fononici immaginari” che a volte saltano fuori con altri modelli e che non hanno significato fisico. Questo ci permette, finalmente, di fare calcoli fononici ad alto rendimento con una precisione all’avanguardia.
Il bello è che il nostro modello non si ferma alla teoria. Abbiamo dimostrato che MACE-MP-MOF0 predice con successo proprietà come l’espansione termica e il modulo di comprimibilità, in ottimo accordo sia con i calcoli DFT più costosi sia con i dati sperimentali disponibili per diversi MOF famosi. Questo apre scenari incredibili per guidare la progettazione di nuovi MOF per applicazioni nell’accumulo di energia e nella termoelettricità.
Come abbiamo addestrato il nostro campione: il dataset
Scegliere il giusto set di addestramento è cruciale quando si parla di machine learning, specialmente con la vastità chimica dei MOF. Oltre a 19 MOF “prototipici” e ampiamente studiati, abbiamo curato altre 108 strutture dal database QMOF, che ne contiene oltre 20.000! Per selezionare questi 108 campioni, ci siamo concentrati su MOF non polarizzati magneticamente con diametri dei pori superiori a 3.6 Å. Abbiamo usato i descrittori MACE stessi (che contengono tantissime informazioni sulle caratteristiche atomiche) per campionare 100 MOF che fossero il più diversi possibile tra loro.
Su questo dataset curato, abbiamo generato dati DFT in vari modi:
- Simulazioni di dinamica molecolare (facendo “muovere” gli atomi a una certa temperatura e pressione) e campionando i frame più significativi.
- Configurazioni “stressate”, cioè espanse e compresse.
- Traiettorie di ottimizzazione geometrica.
In totale, abbiamo ottenuto 4764 punti dati DFT, usati per addestrare e validare il nostro modello. Abbiamo persino creato due versioni, MACE-MP-MOF0 e MACE-MP-MOF0-v2, che differiscono solo per come i dati sono stati divisi, per assicurarci che le prestazioni fossero robuste.
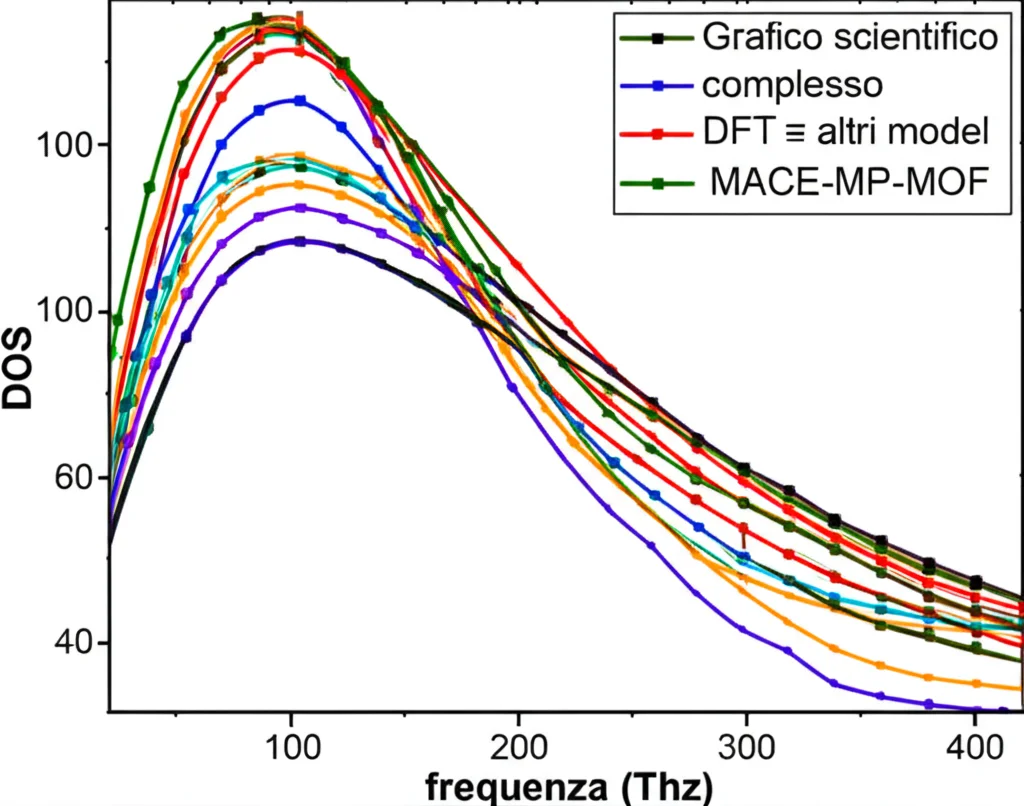
MACE-MP-MOF0 alla prova del nove: i risultati
Il flusso di lavoro con MACE-MP-MOF0 inizia con un rilassamento completo della cella cristallina, senza vincoli di simmetria. Questo è fondamentale per scenari di screening, dove magari non conosciamo la configurazione di simmetria più stabile del MOF. Una volta trovata la struttura di equilibrio (e eliminati eventuali modi immaginari spuri), calcoliamo i fononi usando l’approssimazione quasi-armonica (QHA). Questa approssimazione è un passo avanti rispetto a quella armonica semplice, perché tiene conto di come le frequenze vibrazionali cambino con il volume, e quindi con la temperatura e la pressione, permettendoci di catturare effetti anarmonici importanti.
Abbiamo confrontato MACE-MP-MOF0 con DFT, GFN1-xTB, il modello MACE-MP-0b di partenza, altri MLP specifici per MOF e dati sperimentali. I MOF scelti per questo benchmark sono “celebrità” nel campo: MOF-5 (Zn), UiO-66 (Zr), MOF-74 (Zn) e MIL-53 (Al), tutti diversi per nodi, linker e topologia.
I risultati sono stati entusiasmanti:
- Strutture di equilibrio: Le previsioni di MACE-MP-MOF0 e MACE-MP-MOF0-v2 sono in accordo quasi perfetto con la DFT (deviazione massima dell’1.02%), mentre MACE-MP-0b mostrava deviazioni molto più grandi (fino al 10%). Inoltre, i nostri modelli indovinano il gruppo spaziale corretto, cosa che GFN1-xTB e MACE-MP-0b non sempre facevano, portando a costi computazionali maggiori.
- Forze, energie e stress: Anche qui, precisione stellare rispetto alla DFT, con un miglioramento significativo rispetto a MACE-MP-0b, soprattutto per MOF contenenti metalli come Zn, Al, Mg, Hf e Zr.
- Densità di stati fononici (DOS) e strutture a bande: I nostri modelli MACE-MP-MOF0 sono in ottimo accordo con la DFT su tutto lo spettro di frequenze, anche ben oltre la regione a bassa frequenza. GFN1-xTB e MACE-MP-0b, pur cogliendo alcuni aspetti, fallivano nel descrivere accuratamente MOF prototipici come MOF-5 e UiO-66. È importante sottolineare che abbiamo ottenuto questa accuratezza con un numero di configurazioni di riferimento nel training set da 10 a 100 volte inferiore rispetto ad altri MLP “on-the-fly”, e con costi di generazione dei dati DFT significativamente ridotti!
Non solo teoria: espansione termica negativa e modulo di comprimibilità
Il modulo di comprimibilità (bulk modulus) è un ottimo test per la precisione dei fononi, perché entrambi derivano dalle derivate seconde dell’energia. Ebbene, MACE-MP-MOF0 e MACE-MP-MOF0-v2 non solo catturano qualitativamente l’andamento dei moduli di comprimibilità rispetto a DFT e dati sperimentali, ma riproducono quantitativamente i valori con deviazioni minime. Questo vale anche per MOF “fuori campione”, cioè non inclusi nel dataset di training, come ZIF-4, ZIF-8, UiO-66-Ce, UiO-66-Hf e altri promettenti MOF per la cattura diretta dell’aria. Questo dimostra la trasferibilità e l’accuratezza del nostro modello.
Abbiamo anche testato il modello sull’espansione termica negativa (NTE), un fenomeno affascinante per cui alcuni materiali si restringono quando vengono riscaldati. Per MOF-5 e UiO-66, le previsioni di MACE-MP-MOF0-v2 sono in buon accordo con la DFT e altri modelli computazionali, e catturano qualitativamente gli andamenti sperimentali, come il fatto che UiO-66 mostri una NTE maggiore rispetto a MOF-5.

Perché MACE-MP-MOF0 è così bravo?
L’analisi delle posizioni atomiche nella struttura di equilibrio di MOF-74 ci ha dato una bella dritta. GFN1-xTB e MACE-MP-0b mostravano errori significativi nelle posizioni degli atomi di carbonio e ossigeno dei linker organici, che dominano le vibrazioni a frequenze più alte. I nostri modelli MACE-MP-MOF0, invece, hanno errori da dieci a mille volte inferiori per tutti gli elementi. Questo significa che MACE-MP-MOF0 cattura molto meglio le interazioni covalenti all’interno dei MOF, superando una limitazione di MACE-MP-0b e GFN1-xTB che si concentravano di più sulle interazioni non covalenti. Questo apre le porte all’uso di MACE-MP-MOF0 anche per altri sistemi dove le interazioni covalenti sono dominanti, come i Covalent Organic Frameworks (COF).
Cosa ci riserva il futuro?
MACE-MP-MOF0 è già uno strumento potente, circa il 50% più veloce di MACE-MP-0b e 10 volte più accurato per geometrie, forze, energie e stress. È anche fino al 90% più veloce di GFN1-xTB per MOF grandi. Tuttavia, c’è sempre spazio per migliorare! Ad esempio, potremmo affinare ulteriormente la predizione delle vibrazioni ad altissima frequenza (quelle degli atomi di idrogeno nei linker).
Un’altra area di sviluppo riguarda i MOF con elementi magnetici. Il nostro attuale dataset è composto principalmente da MOF con ioni metallici a “guscio chiuso”, per evitare complicazioni dovute agli stati di spin elettronico. Includere dati di addestramento specifici per configurazioni atomiche con diversi stati di spin potrebbe migliorare le prestazioni per questi sistemi complessi. La bellezza del nostro approccio è che è relativamente facile riparametrizzare il modello per nuove specie chimiche, semplicemente aggiungendo alcune configurazioni di riferimento al dataset DFT.
In conclusione: un passo avanti per la progettazione dei MOF
Spero di avervi trasmesso un po’ del mio entusiasmo! Con MACE-MP-MOF0, abbiamo fatto un bel passo avanti. Ora abbiamo a disposizione uno strumento che ci permette di calcolare proprietà fononiche con qualità ab initio (cioè paragonabile ai metodi più accurati) in modo efficiente e ad alto rendimento. Questo non è solo un traguardo accademico: significa poter accelerare la scoperta e la progettazione di nuovi MOF con proprietà dinamiche complesse, su misura per le applicazioni più disparate, dall’energia pulita alla salute.
La strada è ancora lunga, e l’approssimazione quasi-armonica, sebbene utile, potrebbe non essere sufficiente per descrivere appieno tutti gli effetti anarmonici nei MOF. Ma ogni grande viaggio inizia con un primo passo, e MACE-MP-MOF0 è sicuramente un passo da gigante nella giusta direzione. Continueremo a lavorare per rendere questi modelli sempre più potenti e trasferibili, perché crediamo fermamente che l’unione tra scienza dei materiali e intelligenza artificiale sia la chiave per sbloccare un futuro pieno di innovazioni straordinarie.
Fonte: Springer





