PNET Renale: Viaggio nel Cuore di un Tumore Raro e Aggressivo dalla Cina Nord-Occidentale
Ciao a tutti! Oggi voglio portarvi con me in un viaggio affascinante, anche se un po’ complesso, nel mondo della ricerca oncologica. Parleremo di un nemico piccolo ma incredibilmente tenace: il tumore neuroectodermico primitivo renale, o più semplicemente rPNET. So che il nome suona complicato, ma fidatevi, la storia dietro è degna di attenzione.
Immaginate un tumore così raro da rappresentare meno dell’1% di tutte le neoplasie renali. È talmente sfuggente che diagnosticarlo e trattarlo diventa un vero rompicapo per i medici. Proprio per far luce su questa entità così elusiva, un gruppo di ricercatori ha condotto uno studio retrospettivo multicentrico, analizzando 16 casi provenienti da cinque ospedali nella vasta regione della Cina Nord-Occidentale. Perché proprio lì? Perché anche in aree geografiche specifiche, capire come si comporta una malattia rara può darci indizi preziosi.
Ma cos’è esattamente un PNET?
Prima di tuffarci nei dettagli dello studio, facciamo un passo indietro. I tumori neuroectodermici primitivi (PNET) sono neoplasie aggressive che nascono da cellule embrionali “rimaste indietro”, derivate dalla cresta neurale. Pensateli come piccoli “errori di percorso” durante lo sviluppo fetale. Si dividono in due grandi famiglie:
- I cPNET, che colpiscono il sistema nervoso centrale.
- I pPNET (periferici), che possono spuntare un po’ ovunque: arti, torace, retroperitoneo, pelvi e organi solidi, come il nostro rene (da cui rPNET).
Curiosamente, i pPNET sono strettamente imparentati con un altro tumore osseo noto come Sarcoma di Ewing (EWS), tanto che spesso i termini vengono usati quasi come sinonimi. Hanno caratteristiche cliniche, cellulari e genetiche molto simili.
La Sfida della Diagnosi: Un Nemico Mascherato
Uno dei problemi principali con l’rPNET è che non dà sintomi specifici. Dolore al fianco, una massa palpabile nell’addome, sangue nelle urine (ematuria), gonfiore… sintomi che potrebbero far pensare a tante altre patologie renali, molto più comuni. Anche le immagini radiologiche (TAC, Risonanza) spesso non bastano a distinguerlo con certezza.
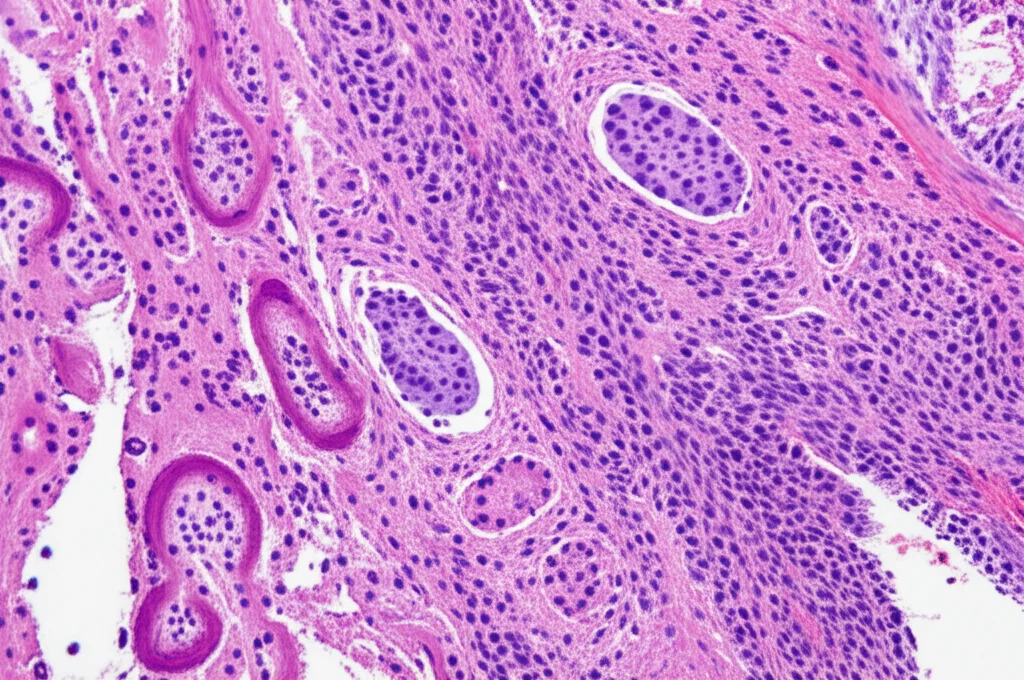
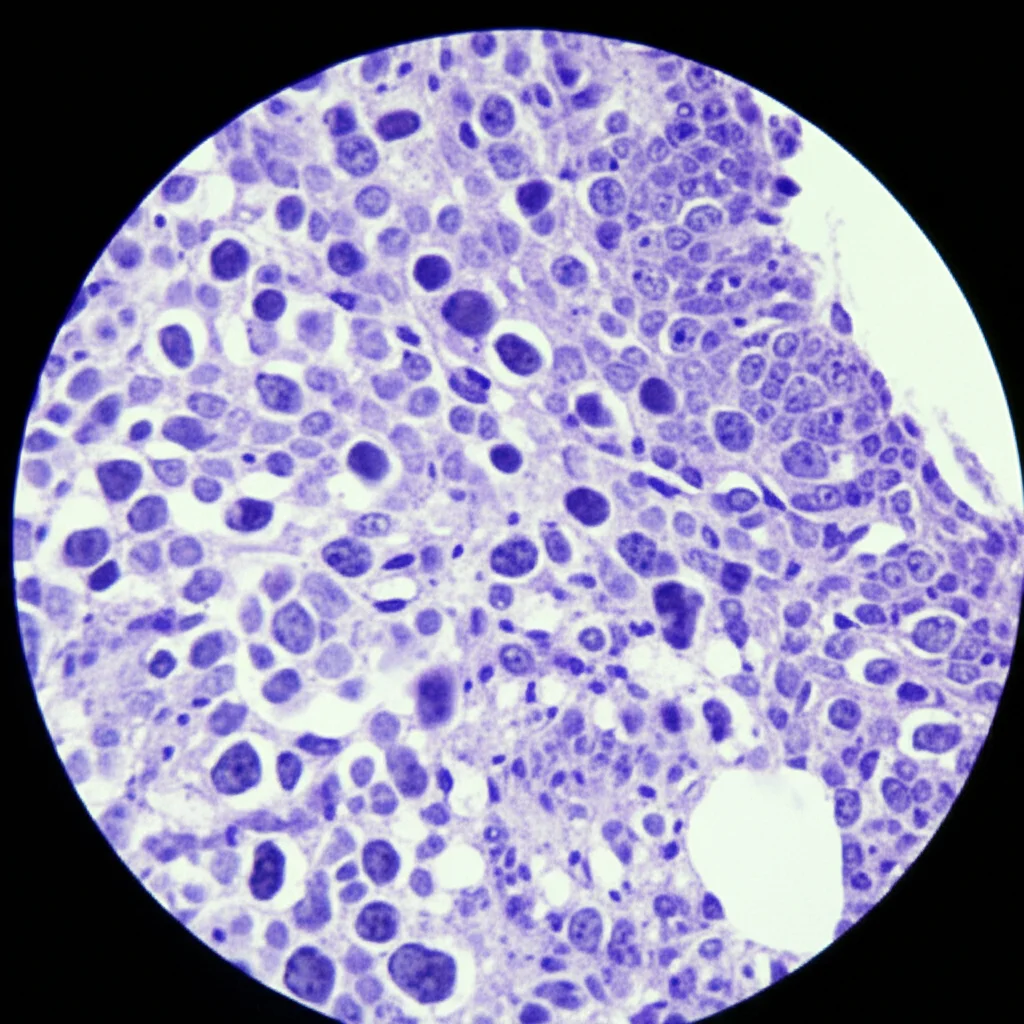
La vera chiave è l’esame istologico, guardare il tumore al microscopio. Lì si vedono le caratteristiche “piccole cellule rotonde blu”, un segno distintivo. Ma non basta! Servono colorazioni speciali (immunoistochimica – IHC) e test genetici molecolari per confermare l’identità del tumore.
Nello studio cinese, ad esempio, tutti i casi mostravano positività diffusa per un marcatore chiamato CD99 e quasi tutti (93.8%) per FLi-1. Inoltre, il 75% esprimeva marcatori neurali come Sinaptofisina (SYN), Cromogranina A (CgA) o Enolasi Neurone-Specifica (NSE), confermando l’origine neuroectodermica. Un altro test fondamentale è la FISH (Ibridazione in Situ Fluorescente) per cercare il riarrangiamento del gene EWSR1, presente in tutti i 16 casi analizzati, un vero “marchio di fabbrica” di questi tumori.
Una curiosità istologica: spesso si cercano delle strutture chiamate “rosette di Homer-Wright” o pseudorosette. Nello studio, erano presenti nel 56.3% dei casi. Ma attenzione: la loro assenza non esclude la diagnosi! Questo ci dice quanto sia eterogeneo questo tumore.
Lo Studio Cinese: Cosa Abbiamo Imparato dai 16 Casi?
Questo studio, seppur basato su un numero limitato di pazienti (16, un numero comunque significativo per una malattia così rara), ci ha dato informazioni preziose:
- Età e Sesso: L’età media alla diagnosi era di 35.4 anni (mediana 39), con un range da 15 a 56 anni. Questo è interessante perché studi americani riportano un’età media più bassa (26 anni). Forse ci sono differenze regionali? La maggioranza erano uomini (10 su 16), ma il sesso non sembrava influenzare la prognosi. L’età più avanzata, invece, è risultata correlata a una sopravvivenza peggiore nell’analisi preliminare.
- Presentazione Clinica: I sintomi più comuni erano dolore al fianco (56.3%), massa addominale (50%), distensione addominale (31.3%) ed ematuria (6.3%). Un paziente era addirittura asintomatico. I tumori erano spesso grandi (diametro medio 8.5 cm).
- Stadio e Metastasi: Purtroppo, molti pazienti arrivavano alla diagnosi con malattia già avanzata: il 50% aveva malattia localmente avanzata (stadio III) e il 31.3% aveva già metastasi a distanza (stadio IV). Il 50% aveva un trombo neoplastico nella vena cava e il 25% metastasi ai linfonodi. Durante il follow-up, oltre la metà dei pazienti (56.3%) ha avuto recidive o metastasi, principalmente alle ossa, polmoni e fegato.
Come si Curano (o si Tenta di Curare) gli rPNET?
Non esiste un protocollo standardizzato, il che complica le cose. L’approccio principale è la chirurgia (nefrectomia radicale o parziale), spesso seguita da chemioterapia (usando regimi come VAC – vincristina, doxorubicina, ciclofosfamide – o IE – ifosfamide, etoposide) e/o radioterapia.
Nello studio, 11 pazienti su 16 hanno subito chirurgia radicale. Nove hanno ricevuto chemioterapia adiuvante, ma due hanno comunque avuto progressione di malattia. Quattro hanno fatto radioterapia.
Una nota interessante riguarda la terapia anti-angiogenica. Tre pazienti hanno ricevuto Anlotinib, un farmaco che blocca la formazione di nuovi vasi sanguigni che nutrono il tumore. Ebbene, tutti e tre hanno risposto bene (2 risposte complete, 1 parziale), e l’analisi ha suggerito che l’uso di questi farmaci fosse associato a una sopravvivenza migliore. È un dato preliminare su pochissimi pazienti, ma apre una porta interessante per future ricerche, dato che questi tumori sono molto vascolarizzati.
Marcatori Molecolari: La Chiave per Capire la Prognosi?
Qui la faccenda si fa ancora più intrigante. I ricercatori hanno analizzato l’espressione di alcune proteine chiave per capire se potessero predire l’andamento della malattia.
- P53: Un famoso gene “guardiano del genoma”. La sua mutazione è stata trovata in molti casi (anche se la definizione di positività nello studio è un po’ complessa, parlando di tipo “mutante” nel 62.5%), ma sorprendentemente, non è risultata significativamente correlata alla sopravvivenza in questa analisi. Forse il campione era troppo piccolo, o forse il suo ruolo è più complesso.
- Ki-67: Questo è un indice di proliferazione, ci dice quanto velocemente le cellule tumorali si moltiplicano. Qui i risultati sono stati chiari: un Ki-67 alto (superiore al 40% nello studio) era un fattore prognostico negativo indipendente. I pazienti con Ki-67 alto avevano una sopravvivenza mediana di soli 12 mesi, contro i 20 mesi di quelli con Ki-67 basso.
- BCL-2: Questa proteina è nota per essere anti-apoptotica, cioè impedisce alle cellule di andare incontro a morte programmata. Normalmente, ci si aspetterebbe che la sua presenza sia un fattore negativo, perché rende le cellule tumorali più resistenti. E invece, tenetevi forte: in questo studio, l’espressione negativa di BCL-2 è risultata un fattore prognostico negativo indipendente! I pazienti senza BCL-2 avevano una sopravvivenza mediana di 10 mesi, contro i 24 mesi di quelli con BCL-2 positivo. Questo risultato è controintuitivo e molto interessante. Gli autori ipotizzano che BCL-2 possa avere ruoli complessi nel microambiente tumorale o nell’equilibrio con altre proteine pro-apoptotiche, forse modulando la risposta immunitaria o infiammatoria. È un dato che necessita assolutamente di ulteriori conferme e studi, soprattutto se si pensa a terapie che mirano a inibire BCL-2!
Sopravvivenza e Prospettive Future
I numeri sulla sopravvivenza purtroppo confermano l’aggressività dell’rPNET: la sopravvivenza globale mediana è stata di 14 mesi. A 1 anno era vivo il 56.3% dei pazienti, a 3 anni solo il 31.3%, e a 5 anni nessuno.
L’analisi multivariata ha confermato che i due fattori indipendenti più importanti per la prognosi erano l’indice Ki-67 (più alto è, peggio è) e l’espressione di BCL-2 (la sua assenza è risultata peggiorativa).

Certo, lo studio ha i suoi limiti: il campione piccolo, la natura retrospettiva, la possibile influenza di fattori regionali specifici della Cina Nord-Occidentale (come l’età più alta alla diagnosi), la variabilità dei trattamenti. Non hanno potuto analizzare la sopravvivenza libera da progressione (PFS) in modo significativo.
Tuttavia, ci lascia messaggi importanti:
- L’rPNET è una sfida diagnostica e terapeutica enorme.
- La diagnosi richiede un approccio integrato: istologia, immunoistochimica (CD99, FLi-1, marcatori neurali) e genetica molecolare (EWSR1). L’assenza di rosette non esclude la diagnosi.
- Marcatori come Ki-67 e BCL-2 sembrano avere un ruolo prognostico cruciale, anche se il ruolo protettivo di BCL-2 necessita di conferme.
- C’è un disperato bisogno di studi più ampi, prospettici e multicentrici per definire meglio la prognosi, standardizzare i trattamenti e testare nuove terapie, come quelle anti-angiogeniche o potenzialmente mirate ai meccanismi molecolari identificati.
Questo studio, pur con le sue limitazioni, è un tassello prezioso che ci aiuta a conoscere meglio questo raro tumore, specialmente in un contesto geografico specifico. Ci ricorda quanto sia fondamentale la ricerca, anche sulle malattie rare, per poter offrire un giorno speranze concrete ai pazienti che ne sono affetti.
Fonte: Springer





