Peptidi Contro il Cancro: La Mia Caccia Digitale all’Inibitore Perfetto di ACSS2
Ciao a tutti! Oggi voglio portarvi con me in un’avventura affascinante nel mondo della ricerca sul cancro, un viaggio che unisce biologia, chimica e la potenza incredibile dei computer. Parleremo di un “cattivo” molecolare chiamato ACSS2 e di come sto cercando, insieme al mio team virtuale (i computer!), di progettare un’arma specifica per neutralizzarlo: un inibitore peptidico. Sembra complicato? Tranquilli, vi guiderò passo passo in questa frontiera della lotta contro il cancro.
Chi è ACSS2 e Perché Dovremmo Preoccuparcene?
Immaginate le cellule tumorali come piccole fabbriche impazzite che hanno bisogno di energia e materiali per crescere senza controllo. L’Acetil-CoA Sintetasi 2 (ACSS2) è un enzima, una sorta di operaio specializzato, che gioca un ruolo cruciale in queste fabbriche. Converte una molecola semplice, l’acetato, in acetil-CoA, un mattone fondamentale per il metabolismo energetico e non solo.
La cosa interessante è che ACSS2 non se ne sta buono buono solo nel citoplasma (la “fabbrica” principale della cellula), ma può anche traslocare nel nucleo, il “centro di comando”. Qui, produce acetil-CoA localmente, influenzando l’acetilazione degli istoni. Pensate agli istoni come a rocchetti attorno ai quali si avvolge il DNA: modificarli significa accendere o spegnere geni specifici. ACSS2, quindi, non solo fornisce energia, ma regola anche l’espressione genica, la divisione cellulare e persino la capacità delle cellule tumorali di adattarsi a condizioni difficili, come la mancanza di ossigeno (ipossia) tipica dell’ambiente tumorale (TME).
Studi hanno dimostrato che molti tumori (glioblastoma, cancro al seno, al fegato, alla prostata…) presentano livelli elevati di ACSS2. Più ACSS2 c’è, spesso peggiore è la prognosi per il paziente. Bloccare ACSS2, quindi, sembra una strategia promettente per fermare la crescita tumorale e magari rendere i tumori più sensibili ad altre terapie.
La Sfida: Trovare l’Inibitore Giusto
Ok, abbiamo il bersaglio: ACSS2. Ora serve l’arma. Esistono già alcune piccole molecole in studio, come il VY-3-135 o il più recente AD-5584, e una (MTB-9655) è persino arrivata ai trial clinici di Fase I. Tuttavia, la ricerca di inibitori è sempre una sfida. Bisogna garantirne la specificità (che colpiscano solo ACSS2 e non altri enzimi simili, come ACSS1 o ACSS3, per evitare effetti collaterali), l’efficacia e capire bene come si comportano nel corpo (farmacocinetica).
Qui entrano in gioco i peptidi. I peptidi sono piccole catene di amminoacidi, i mattoni delle proteine. Rispetto alle piccole molecole tradizionali, offrono vantaggi interessanti:
- Possono avere superfici di interazione più grandi, adattandosi meglio a bersagli proteici complessi o “piatti”, difficili da colpire con molecole piccole.
- Possono essere progettati per essere estremamente selettivi e potenti.
- Generalmente, hanno un buon profilo di sicurezza.
Certo, anche i peptidi hanno le loro “gatte da pelare”, come la stabilità nel corpo o la capacità di entrare nelle cellule. Ma la chimica moderna ci offre trucchi per migliorarli: ciclizzazione, uso di amminoacidi non naturali (D-amminoacidi), N-metilazione… Insomma, sono candidati affascinanti!
La Nostra Strategia Computazionale: Progettare a Tavolino
E se potessimo progettare l’inibitore peptidico perfetto al computer, prima ancora di sintetizzarlo in laboratorio? È qui che inizia la nostra avventura digitale! Abbiamo usato un approccio in silico, sfruttando la potenza di calcolo per simulare e prevedere.
Il punto di partenza? Il sito di legame del nucleotide di ACSS2. Sappiamo che gli inibitori noti si legano lì, competendo con l’ATP (la “benzina” energetica della cellula) e bloccando l’attività dell’enzima. Abbiamo identificato la sequenza di amminoacidi chiave di questo sito in ACSS2 (GEPDTYWQ), basandoci su studi precedenti e confrontando la struttura cristallina di ACSS2 da Candida albicans (un lievito, PDB ID: 8W0D, dato che quella umana non è ancora disponibile) con un modello 3D predittivo di quella umana (AlphaFold). Sorprendentemente, sono molto simili, specialmente nel sito che ci interessa!
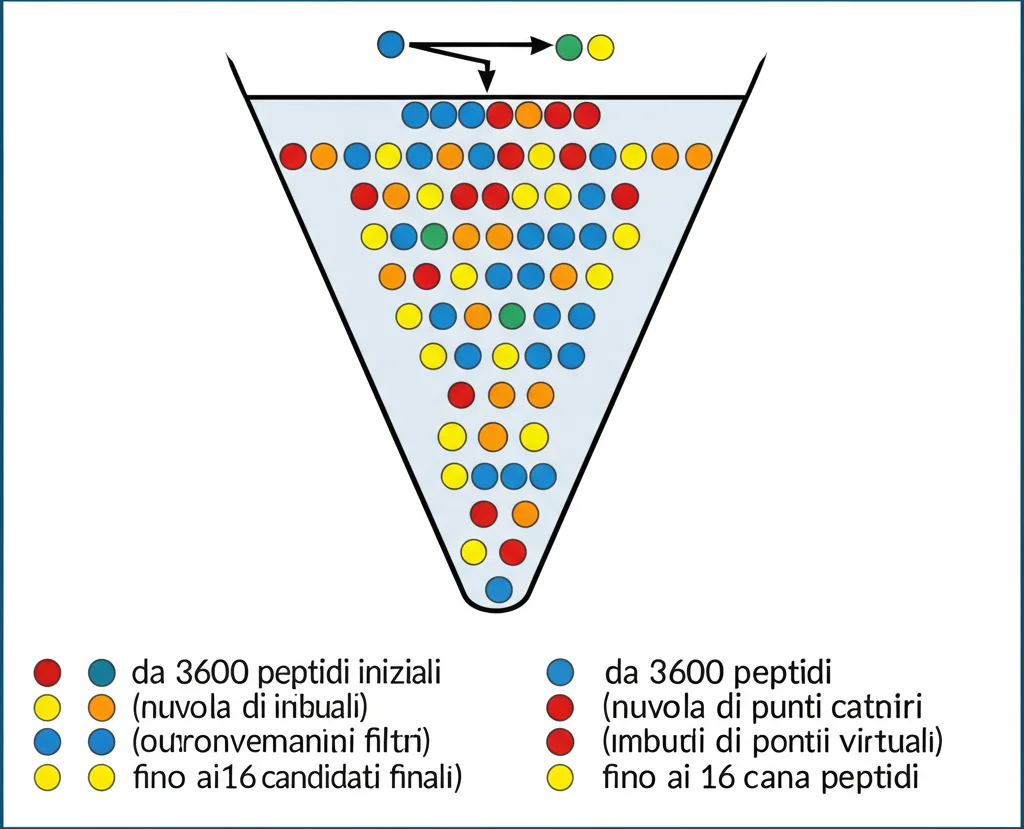
Poi, abbiamo usato uno strumento chiamato Peptide Combination Generator. Abbiamo classificato i 20 amminoacidi naturali in 6 gruppi basati sulle loro proprietà fisico-chimiche (acidi, basici, idrofobici, aromatici, polari, speciali). Partendo dalla sequenza GEPDTYWQ, il generatore ha creato tutte le possibili combinazioni “complementari”, sostituendo ogni amminoacido con uno dello stesso gruppo. Risultato? Una libreria virtuale di ben 3600 sequenze peptidiche combinate (PCS)!
Filtrare, Filtrare, Filtrare: La Selezione dei Campioni
Avere 3600 candidati è fantastico, ma dobbiamo trovare i migliori. È iniziata una fase di screening rigorosissima, tutta al computer:
- Tossicità: Abbiamo usato ToxinPred, un tool basato su machine learning (Support Vector Machine – SVM), per predire la potenziale tossicità di ogni peptide. Abbiamo tenuto solo quelli con un punteggio SVM > -0.30 (meno probabilità di essere tossici). Da 3600 a 172 PCS.
- Proprietà “Drug-like”: Abbiamo filtrato ulteriormente basandoci su:
- Peso molecolare: inferiore a 900 Dalton (un buon compromesso per un peptide).
- Anfipaticità: > 0.30 (importante per interagire con le membrane). Da 172 a 88 PCS.
- Idrofobicità: >= -0.2 (necessaria per interagire con le tasche idrofobiche del bersaglio, ma non troppo alta per evitare aggregazione). Da 88 a 19 PCS.
- Tendenza all’Aggregazione: Un peptide che si aggrega è inutile. Abbiamo usato AGGRESCAN per calcolare la propensione all’aggregazione in vivo (punteggio Na4vSS). Abbiamo tenuto solo quelli con Na4vSS < -57. Da 19 ai nostri Top 16 PCS finalisti!
Questo processo ci ha permesso di selezionare peptidi con un profilo promettente: potenzialmente non tossici, con buone proprietà farmacologiche e basso rischio di aggregazione.
Dare una Forma ai Candidati: Predizione Strutturale e Docking
Ora avevamo 16 sequenze promettenti, ma come si dispongono nello spazio? La struttura 3D è fondamentale per capire come interagiranno con ACSS2. Abbiamo usato PEP-FOLD4, un server avanzato che predice la struttura tridimensionale dei peptidi de novo (da zero), basandosi sulla sequenza e su un campo di forze specifico (sOPEP). Abbiamo ottenuto i modelli 3D più probabili ed energeticamente stabili per ciascuno dei 16 peptidi.
Il passo successivo? Vedere come questi peptidi si “incastrano” nel sito attivo di ACSS2. È il momento del docking molecolare! Abbiamo usato HADDOCK, un software sofisticato che simula l’interazione tra proteine e peptidi, tenendo conto della loro flessibilità. Abbiamo definito i residui del sito attivo di ACSS2 e lasciato che HADDOCK trovasse la migliore “posa” di legame per ogni peptide.
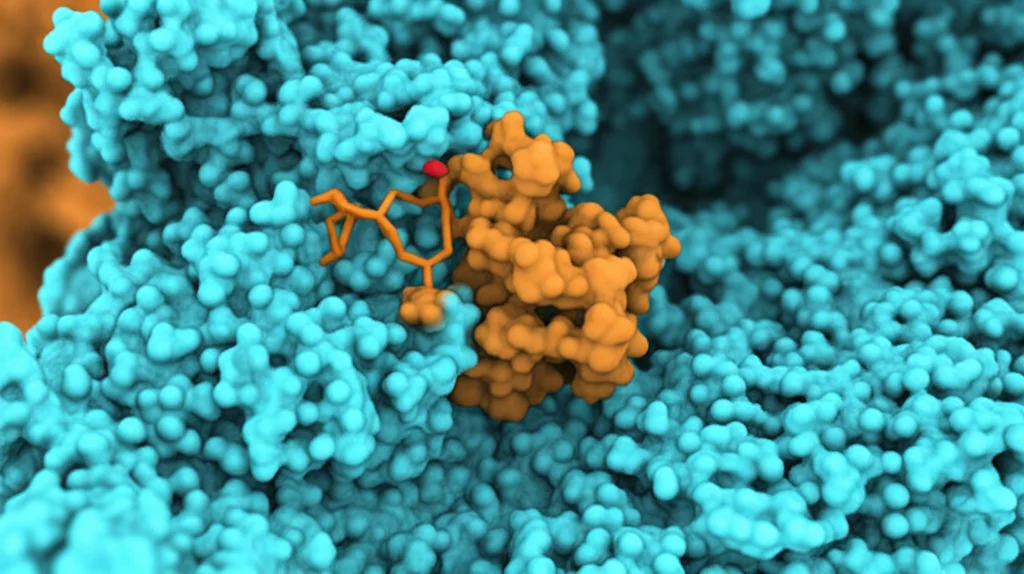
I risultati sono stati elettrizzanti! Tutti i 16 peptidi hanno mostrato affinità di legame (misurata dal punteggio HADDOCK) significativamente superiori a quella dell’inibitore di riferimento AD5584. Il migliore tra i migliori? Un peptide che abbiamo chiamato Pep16, con un punteggio quasi doppio rispetto al riferimento (-91.1 vs -53.7 kcal/mol)! Pep16 sembrava legarsi non solo al sito dell’ATP ma estendersi anche verso il sito di legame del Coenzima A (CoA), un altro attore nella reazione catalizzata da ACSS2.
Pep16 Sotto la Lente: Simulazioni di Dinamica Molecolare
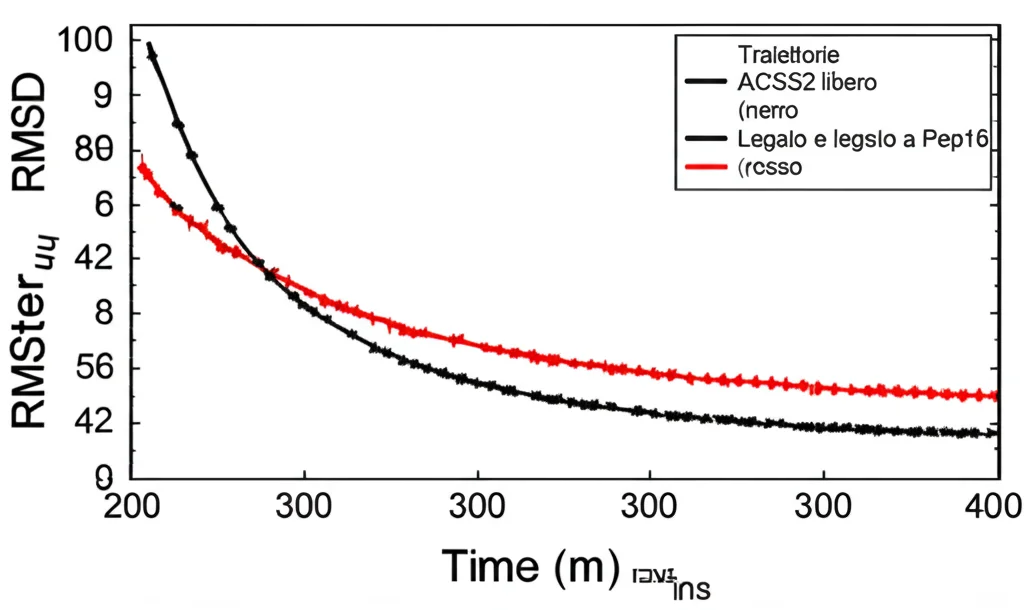
Avere un buon punteggio di docking è ottimo, ma è una fotografia statica. Volevamo vedere il film! Abbiamo quindi eseguito simulazioni di dinamica molecolare (MD) per 300 nanosecondi (un tempo lunghissimo su scala molecolare!) usando il pacchetto software AMBER. Abbiamo simulato tre sistemi: ACSS2 da solo (apo), ACSS2 legato a AD5584 e ACSS2 legato al nostro campione, Pep16.
Le simulazioni MD ci permettono di osservare come le molecole si muovono, vibrano e interagiscono nel tempo in un ambiente acquoso simulato, simile a quello cellulare. Abbiamo analizzato vari parametri:
- Stabilità (RMSD): Pep16 sembrava indurre leggere fluttuazioni nella struttura generale di ACSS2, rendendola un po’ meno stabile rispetto alla forma libera.
- Flessibilità (RMSF): Le simulazioni hanno mostrato che ACSS2 legato a Pep16 era leggermente più flessibile.
- Compattezza (RoG) e Area Superficiale Accessibile al Solvente (SASA): Il complesso Pep16-ACSS2 tendeva ad essere leggermente meno compatto ma più “ripiegato” (minore SASA) rispetto all’enzima libero.
- Movimenti Complessivi (PCA): L’analisi delle componenti principali ha confermato che Pep16 induceva maggiori cambiamenti conformazionali in ACSS2 rispetto alla forma libera.
Questi dati suggeriscono che Pep16 non si limita a “sedersi” nel sito attivo, ma induce cambiamenti dinamici nell’intera struttura dell’enzima.
L’Abbraccio Molecolare: Energia di Legame e Interazioni Chiave
Ma quanto è forte questo legame? E quali forze lo tengono insieme? Abbiamo usato la tecnica MM/GBSA (Molecular Mechanics/Generalized Born Surface Area) per calcolare l’energia libera di legame (BFE). Ancora una volta, Pep16 ha superato AD5584, mostrando un’energia di legame più favorevole.
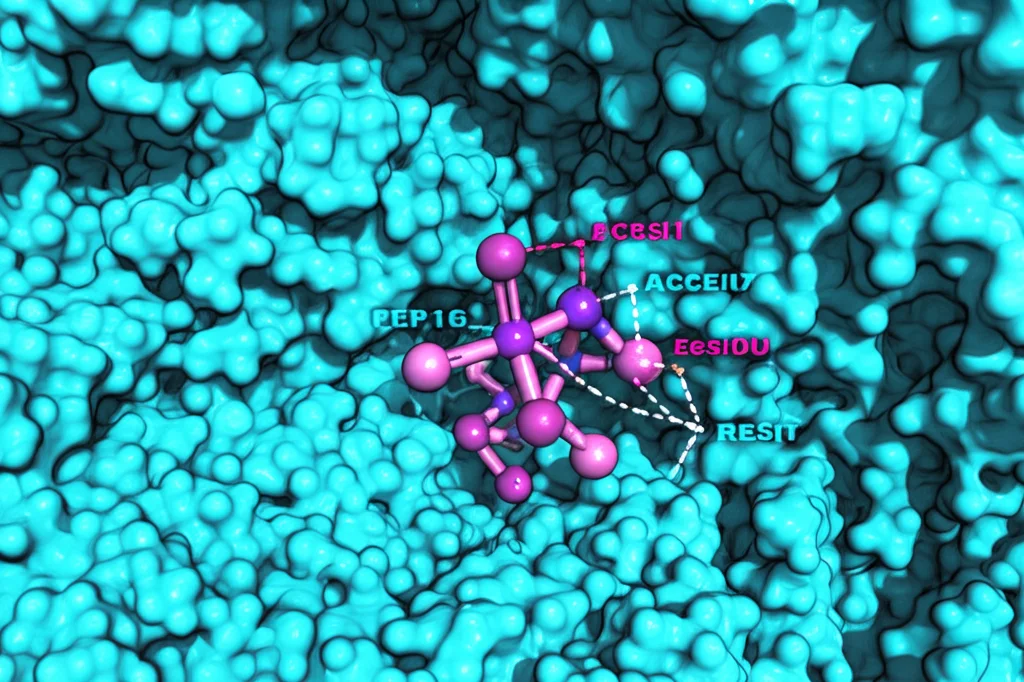
Analizzando più a fondo (con PRED analysis), abbiamo scoperto che le interazioni elettrostatiche (attrazioni tra cariche opposte) erano la forza dominante nel tenere Pep16 legato ad ACSS2. Residui specifici di ACSS2, carichi positivamente (Arginine: ARG 373, ARG 526, ARG 628, ARG 631; Lisina: LYS 632), giocavano un ruolo chiave nell’attrarre Pep16. Inoltre, si formavano legami idrogeno e altre interazioni non covalenti. Pep16 occupava una tasca di legame ampia, interagendo con ben 34 residui di ACSS2 durante la simulazione!
L’Effetto Bloccante: Come Pep16 Potrebbe Funzionare
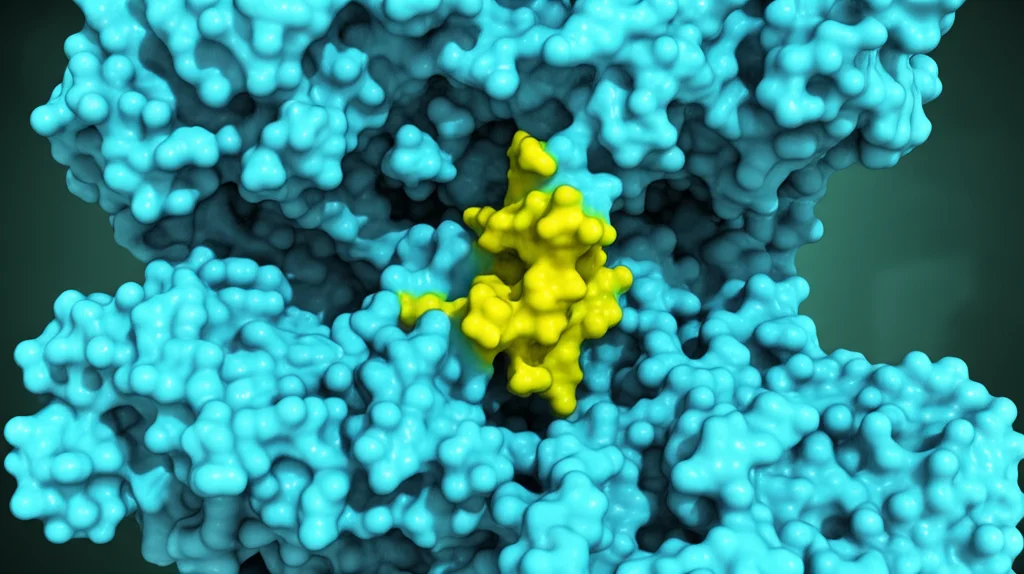
La cosa più intrigante è stata osservare cosa succedeva specificamente ai residui della tasca di legame di ACSS2 quando Pep16 era presente. Contrariamente alla tendenza generale dell’enzima, la tasca di legame diventava più stabile, più rigida e più compatta! Le simulazioni (RMSD, RMSF, RoG, SASA specifici per la tasca e analisi DCCM) hanno confermato questo “irrigidimento” locale. Anche l’analisi della struttura secondaria (DSSP) ha mostrato cambiamenti specifici indotti da Pep16, con un aumento delle strutture alfa-elica nella regione legata, suggerendo una conformazione più ordinata e rigida.
La nostra ipotesi? Pep16 sembra “bloccare” il sito di legame del nucleotide in una conformazione rigida e compatta. Questo potrebbe fisicamente impedire all’ATP di legarsi o di posizionarsi correttamente per la reazione catalitica, bloccando di fatto l’attività di ACSS2. È come mettere una chiave sagomata male in una serratura: non solo non apre, ma impedisce anche alla chiave giusta di entrare.
La Strada è Ancora Lunga, Ma Promettente
Ovviamente, questo è uno studio computazionale. Pep16 è, per ora, una promessa digitale. Il prossimo passo fondamentale sarà sintetizzarlo in laboratorio e testarlo in vitro e poi in vivo per confermare la sua efficacia e sicurezza. Bisognerà confrontarlo direttamente con gli inibitori esistenti.
Inoltre, possiamo già pensare a come migliorare ulteriormente Pep16. Potremmo modificarlo chimicamente per aumentarne la stabilità o la capacità di entrare nelle cellule tumorali. Potremmo incapsularlo in nanoparticelle per un rilascio mirato. Potremmo persino usarlo in combinazione con altre terapie antitumorali per un attacco sinergico.
Conclusione: Un Nuovo Orizzonte Digitale
Questa ricerca, per me, è la dimostrazione di come gli approcci computazionali stiano rivoluzionando la scoperta di farmaci. Siamo partiti da un bersaglio biologico, abbiamo generato migliaia di candidati virtuali, li abbiamo selezionati con criteri rigorosi e abbiamo studiato in dettaglio l’interazione del migliore, Pep16, con il suo bersaglio a livello atomico.
Pep16 rappresenta un potenziale nuovo inibitore specifico per ACSS2, progettato grazie a questa potente metodologia computazionale. Se le verifiche sperimentali confermeranno le nostre previsioni, potrebbe aprire nuove strade per trattare i tumori che dipendono da questo enzima. Al di là del singolo peptide, credo fermamente che questo tipo di approccio sistematico sarà uno strumento indispensabile per la scoperta futura di farmaci peptidici, aprendo nuovi orizzonti nella lotta contro il cancro e altre malattie. L’avventura continua!
Fonte: Springer