P53 Sotto Attacco: Come la Sua Degradazione Scatena il Cancro al Colon-Retto
Ciao a tutti! Oggi voglio parlarvi di qualcosa che mi appassiona tantissimo e che sta cambiando il modo in cui vediamo l’insorgenza di uno dei tumori più diffusi: il cancro del colon-retto (CRC). Per anni, abbiamo seguito un modello ben definito, quasi una “storia” molecolare, proposta da Fearon e Vogelstein. Questa storia diceva che tutto inizia con problemi alla proteina APC, che fa partire la via di segnalazione WNT, portando alla formazione di polipi. Poi, passo dopo passo, altre mutazioni (come quelle in KRAS) spingono questi polipi a diventare più aggressivi, fino alla perdita finale del “guardiano del genoma”, la proteina p53, che segna il passaggio ad adenocarcinoma maligno.
Il Vecchio Modello e le Domande Aperte
Questo modello è stato fondamentale, non fraintendetemi. Ci ha dato una mappa preziosa. Ma come in tutte le grandi storie, c’erano dei dettagli che non quadravano del tutto, specialmente riguardo alle primissime fasi. Cosa scatena davvero la trasformazione iniziale? E come fanno le cellule ad accumulare quel carico di mutazioni “giuste” per passare da un polipo innocuo a un cancro aggressivo? Queste erano le domande che mi (e molti altri ricercatori) tenevano sveglio la notte. Sembrava mancasse un pezzo del puzzle, un meccanismo che collegasse l’inizio con la progressione più maligna. E se vi dicessi che la perdita di p53, tradizionalmente vista come un evento tardivo, potesse in realtà giocare un ruolo chiave *fin dall’inizio*?
L’Indizio Inaspettato: Entra in Scena URI
Ragazzi, preparatevi, perché quello che abbiamo scoperto potrebbe riscrivere un capitolo importante. Integrando dati da pazienti umani con esperimenti su modelli murini geneticamente modificati, abbiamo identificato un nuovo attore chiave: una proteina oncogenica chiamata URI. Analizzando database come il TCGA (The Cancer Genome Atlas), abbiamo notato una cosa interessante: anche se il gene URI1 non è spesso amplificato o mutato nel CRC, il suo mRNA è significativamente sovraespresso nei tumori rispetto al tessuto normale adiacente. E non solo nel CRC, ma anche in altri tipi di cancro! Questa sovraespressione di URI correlava negativamente con la sopravvivenza dei pazienti, specialmente in un sottotipo molecolare specifico di CRC (chiamato CMS2), noto per l’attivazione delle vie WNT/MYC. Bingo!
Ma la cosa ancora più intrigante è che alti livelli di URI si trovavano già nelle fasi precoci, come nei polipi adenomatosi umani (le lesioni precancerose) e persino in pazienti con colite ulcerosa, una condizione infiammatoria che predispone al CRC. Era come se URI fosse lì, sulla scena del crimine, fin dai primi momenti.
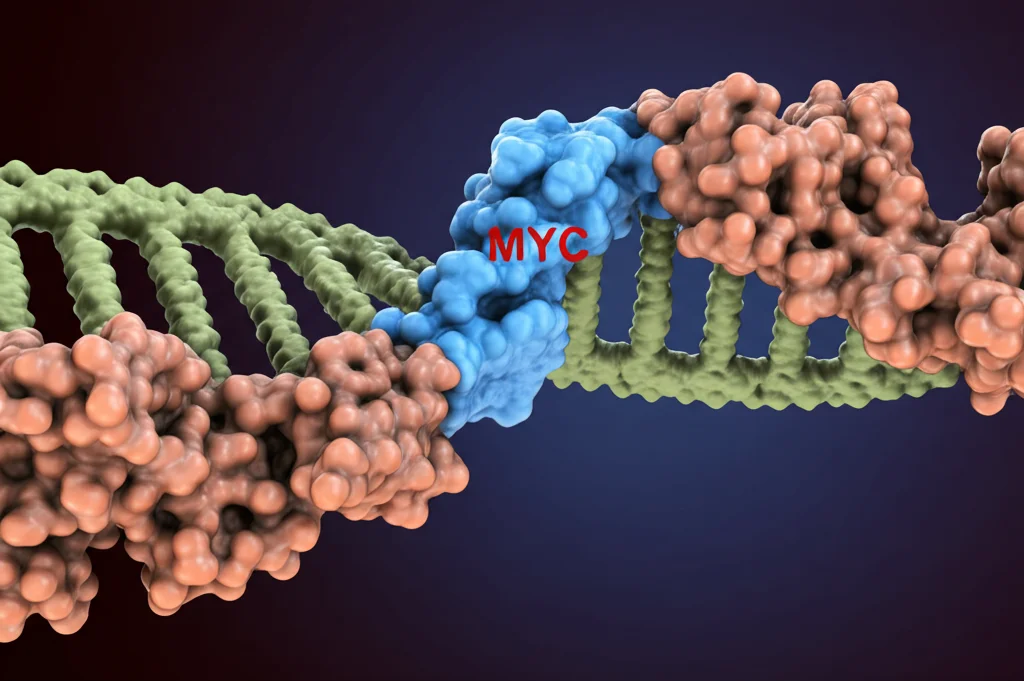
Il Legame Pericoloso: MYC Attiva URI
A questo punto, la domanda era: cosa causa l’aumento di URI? Abbiamo guardato ai sospetti noti nel sottotipo CMS2, e il principale indiziato era MYC. MYC è un potente oncogene, uno dei primi ad essere attivati dopo la perdita di APC nella via WNT. E indovinate un po’? I livelli di mRNA di URI correlavano perfettamente con quelli di MYC nei pazienti con CRC. Non solo, ma analizzando il DNA, abbiamo scoperto che MYC può legarsi direttamente alle regioni regolatrici (promotore ed enhancer) del gene URI1, sia nell’uomo che nei topi. Esperimenti in cellule tumorali umane (RKO CRC) hanno confermato questo legame diretto (ChIP-qPCR) e dimostrato che sovraesprimere MYC aumentava l’attività del promotore di URI, mentre silenziare MYC riduceva i livelli della proteina URI. Era chiaro: MYC, attivato dalla perdita iniziale di APC, ordina direttamente la produzione dell’oncogene URI. Questo legame era il primo passo cruciale.
Il Colpo di Scena: URI Fa Fuori p53!
E qui arriva il bello. Cosa fa URI una volta che è sovraespresso? Abbiamo rivolto la nostra attenzione a p53. Sapevamo da studi precedenti e dai dati umani (ricordate la correlazione tra alti livelli di URI e mutazioni/perdita di TP53?) che poteva esserci un legame. Utilizzando modelli murini dove potevamo eliminare specificamente URI nell’epitelio intestinale (topi Uri(+/Δ)vil), abbiamo visto che la riduzione di URI portava a un *aumento* dei livelli della proteina p53 (ma non del suo mRNA!). Al contrario, sovraesprimendo URI umano nell’intestino dei topi (topi hURI(+/KI)vil), i livelli di p53 crollavano.
Come faceva URI a controllare p53? Non modificava i livelli di MDM2, la famosa proteina E3 ligasi che marca p53 per la distruzione. Esperimenti in cellule CRC umane hanno mostrato che URI riduceva l’emivita di p53 e aumentava la sua ubiquitinazione (il “marchio della morte” per le proteine). La chiave era che URI si legava sia a p53 che a MDM2, formando un complesso a tre. E la cosa pazzesca è che, in esperimenti *in vitro* con proteine purificate, abbiamo dimostrato che URI aumenta direttamente l’attività di MDM2, rendendola più efficiente nel marcare p53 per la degradazione da parte del proteasoma. Sembra quasi un thriller molecolare, vero? URI non si limita a essere presente, ma agisce attivamente per eliminare il guardiano p53.

La Prova Definitiva nei Modelli Murini di CRC
Ora dovevamo dimostrare che questo meccanismo fosse davvero cruciale per l’inizio del tumore nel contesto della perdita di APC. Siamo tornati ai nostri topi modello di CRC (Apc(+/15Δ)vil), che mimano la perdita di APC umana e sviluppano tumori intestinali. Come previsto, questi topi avevano alti livelli di MYC e URI, e… bassi livelli di p53!
Allora abbiamo fatto degli esperimenti decisivi:
- Ripristinare p53: Incrociando questi topi con i “Super p53” (che hanno copie extra del gene p53), abbiamo visto una drastica riduzione del numero e della dimensione dei tumori. Mantenere p53 attivo bloccava l’iniziazione!
- Ridurre URI: Creando topi con perdita di APC e *meno* URI (Apc(+/15Δ)vil;Uri(+/Δ)vil), abbiamo visto che i livelli di p53 si rialzavano e, di conseguenza, i topi sviluppavano molti meno tumori, erano meno gravi e vivevano più a lungo.
- Il ruolo di p53: Ma la prova del nove è stata questa: se nei topi con meno URI (che erano protetti) eliminavamo geneticamente anche p53, *tutti* i benefici della riduzione di URI svanivano! I tumori tornavano numerosi e aggressivi. Questo dimostrava in modo inequivocabile che l’effetto protettivo della riduzione di URI dipendeva totalmente dalla sua capacità di salvare p53 dalla degradazione.
- Aumentare URI: Al contrario, sovraesprimere URI nei topi con perdita di APC peggiorava la situazione, aumentando il numero di lesioni precoci, proprio come faceva l’eliminazione genetica di p53.
Abbiamo ottenuto risultati simili anche in un altro modello, dove attivavamo direttamente β-catenina (il segnale a valle di APC), confermando che l’asse MYC-URI-p53 è centrale quando la via WNT è iperattiva.

E KRAS? Un Attore Secondario (all’Inizio)
Ok, ma che fine fa KRAS in questa nuova storia? Secondo il vecchio modello, le sue mutazioni arrivano dopo APC e prima di p53. Abbiamo testato anche questo. Attivare una mutazione comune di KRAS (KRASV12) nell’intestino dei topi, da sola, non causava tumori. Ma se la attivavamo *insieme* alla perdita di APC, i tumori si formavano, ed erano anche più aggressivi, spostandosi verso il colon (come spesso accade nell’uomo) e dando persino metastasi al fegato se p53 era assente.
Tuttavia, anche in questo modello combinato (APC loss + KRASV12), ridurre URI diminuiva ancora il numero iniziale di tumori, anche se non fermava l’aggressività data da KRAS. Questo conferma che la degradazione di p53 mediata da URI è un evento *iniziale*, che avviene prima e indipendentemente dalle mutazioni di KRAS, le quali sembrano piuttosto responsabili della progressione successiva.
Quando p53 Manca Davvero
È importante notare una cosa: se un tumore nasce *direttamente* dalla perdita genetica di p53, senza che ci sia stata una perdita iniziale di APC, allora l’asse MYC-URI non sembra coinvolto. In topi dove abbiamo eliminato p53 nell’intestino fin dall’inizio, i tumori che si formavano (più tardi e meno frequentemente) non mostravano alti livelli di URI, e modulare URI (su o giù) non cambiava l’incidenza dei tumori. Questo rafforza l’idea che il meccanismo MYC-URI-p53 sia specifico per l’iniziazione guidata dalla via WNT/APC.
Un Nuovo Modello per l’Inizio del CRC
Quindi, cosa ci dice tutto questo? Che dobbiamo aggiornare la nostra “mappa” del CRC! Ecco il nuovo scenario che proponiamo:
- Primo colpo (Hit 1): Perdita di APC (o mutazioni equivalenti) -> Attivazione di β-catenina -> Attivazione di MYC.
- Secondo colpo (Hit 2 – Il nostro contributo!): MYC sovraesprime l’oncogene URI -> URI potenzia l’attività di MDM2 -> p53 viene degradato via proteasoma.
- Conseguenza: La perdita funzionale di p53 (anche se il gene è ancora lì!) permette alle cellule di proliferare senza controllo, accumulare stress replicativo e danno al DNA. Questo crea un ambiente instabile che facilita l’acquisizione di ulteriori mutazioni (transizionali), come quelle in KRAS.
- Progressione: Mutazioni successive (KRAS, perdita genetica di p53, ecc.) spingono l’adenoma verso l’adenocarcinoma aggressivo e metastatico.
Questa degradazione precoce di p53 spiega anche meglio perché l’invecchiamento è un fattore di rischio: con l’età, i meccanismi di controllo come p53 possono indebolirsi, rendendo più facile per l’asse MYC-URI prendere il sopravvento dopo un “primo colpo” su APC.
Implicazioni e Futuro
Questa scoperta è entusiasmante perché non solo ci dà una comprensione più profonda di come inizia il CRC, ma apre anche nuove strade. Se la degradazione di p53 mediata da URI è così cruciale all’inizio, allora URI diventa un bersaglio terapeutico potenzialmente potentissimo per la *prevenzione* o il trattamento precoce del CRC, specialmente in quel sottogruppo CMS2 guidato da WNT/MYC. Immaginate farmaci che bloccano l’interazione URI-MDM2 o l’attività di URI: potrebbero impedire a p53 di essere degradato e fermare il tumore sul nascere!
Certo, c’è ancora lavoro da fare. Dobbiamo capire se URI ha anche altri ruoli oncogenici nelle fasi più avanzate (i nostri dati suggeriscono di sì, forse indipendenti da p53). Ma aver identificato questo meccanismo iniziale, questo “tallone d’Achille” del tumore nascente, è un passo avanti enorme. Spero che questa nuova prospettiva ci aiuti a sviluppare strategie migliori contro questa malattia devastante.
Fonte: Springer





