Sordità Ereditaria: Ho Scoperto Perché un Gene ‘Difettoso’ Ci Rende Sordi (Grazie ai Topi!)
Ciao a tutti! Oggi voglio raccontarvi una storia affascinante che arriva direttamente dal mio laboratorio, una di quelle avventure scientifiche che ti fanno battere il cuore. Parliamo di udito, o meglio, di quando l’udito viene a mancare a causa di un piccolo “errore” nel nostro DNA. La sordità ereditaria è un puzzle incredibilmente complesso, con tantissimi pezzi diversi – pensate che ad oggi conosciamo più di 150 geni legati alla sordità non sindromica!
Un Gene Sotto la Lente: SLC12A2
Al centro della nostra storia c’è un gene chiamato SLC12A2. Forse il nome non vi dice molto, ma è importantissimo. Questo gene è stato collegato a una forma di sordità ereditaria autosomica dominante chiamata DFNA78. La cosa curiosa è che tutte le varianti “difettose” di questo gene trovate finora nei pazienti colpiscono una specifica regione, l’esone 21.
Ma cosa fa di così importante questo SLC12A2? Produce una proteina chiamata NKCC1, un co-trasportatore che lavora come un instancabile operaio nelle nostre cellule, aiutando a regolare la pressione osmotica interna. Nell’orecchio interno, in particolare nella coclea (la nostra “chiocciola” uditiva), NKCC1 è fondamentale per produrre l’endolinfa, un liquido speciale ricco di potassio che riempie una parte della coclea chiamata Scala Media (SM) ed è essenziale per far funzionare le cellule sensoriali dell’udito. Immaginate la coclea divisa in “stanze”: la Scala Media con l’endolinfa, e altre due (Scala Vestibuli e Scala Tympani) piene di perilinfa. La proteina NKCC1 si trova principalmente nelle cellule marginali della stria vascolare (StV), una specie di “fabbrica” specializzata nella parete laterale della coclea, proprio dove si produce l’endolinfa.
Il Mistero dell’Esone 21 e la DFNA78
Come dicevo, nei pazienti con DFNA78, le mutazioni colpiscono sempre l’esone 21 di SLC12A2. Studi in vitro hanno mostrato che se questo esone manca o è alterato, la proteina NKCC1 non riesce più a trasportare correttamente gli ioni (come il Cloro). Ma c’era un dubbio: la sordità è causata solo da una ridotta attività di trasporto (aploinsufficienza), o c’è dell’altro? La faccenda si complica perché esistono persone con varianti di SLC12A2 che dovrebbero “spegnere” il gene, ma non mostrano sintomi evidenti (ad esempio, i genitori di pazienti con la sindrome di Kilquist, una malattia recessiva legata a SLC12A2).
Questo ci ha fatto pensare: forse la proteina senza la regione codificata dall’esone 21 (che chiameremo Δex21) non solo funziona male come trasportatore, ma acquisisce anche qualche funzione anomala o perde un ruolo cruciale. Si era ipotizzato che l’esone 21 fosse importante per indirizzare la proteina NKCC1 al posto giusto nella cellula (la membrana basolaterale), ma i risultati degli studi erano contraddittori. Insomma, serviva un modello animale per capirci qualcosa di più! I topi knockout per Slc12a2 (cioè senza il gene) non erano adatti, perché mostrano una riduzione gravissima dell’endolinfa fin da subito, un quadro diverso dalla DFNA78.
Creare il Modello: Nascono i Topi Em1 ed Em2
Ed eccoci al cuore del nostro lavoro. Abbiamo usato la potente tecnica CRISPR/Cas9 per “modificare” il gene Slc12a2 nei topi, cercando di replicare una mutazione trovata nei pazienti DFNA78 che causa l’omissione dell’esone 21. Abbiamo generato due ceppi di topi:
- Slc12a2Em1: con una mutazione puntiforme (c.2912-2 A>G) nel sito di splicing dell’esone 21, che pensavamo fosse equivalente a quella umana.
- Slc12a2Em2: con una piccola delezione di 6 basi (c.2912-4_2913del) sempre vicino all’esone 21.
Ci siamo concentrati soprattutto sul modello Em2. E qui le cose si sono fatte davvero interessanti!
Il Topo Em2: Un Modello Fedele (e Sordo) della DFNA78
Analizzando l’RNA dei topi Slc12a2Em2/Em2 (omozigoti, cioè con entrambe le copie del gene mutate), abbiamo confermato che nei loro trascritti mancava completamente l’esone 21. Proprio come ipotizzato! E le conseguenze? Drammatiche per l’udito. A 4 settimane di vita, questi topolini mostravano comportamenti tipici di un disturbo dell’orecchio interno (giravano in tondo, scuotevano la testa) e i test uditivi (i potenziali evocati uditivi del tronco encefalico, o ABR) hanno rivelato una sordità profonda: non rispondevano a nessun suono testato, anche a volumi altissimi. Questo quadro assomiglia molto alla sordità grave o profonda osservata nei pazienti con DFNA78.
Anche i topi eterozigoti (Slc12a2Em2/+), con una sola copia mutata, hanno mostrato qualcosa: le femmine, in particolare, avevano soglie uditive leggermente più alte a certe frequenze (16 kHz e 32 kHz) rispetto ai topi normali. Un effetto più lieve, ma significativo.
Dentro la Coclea del Topo Em2: Cosa Non Funziona?
Siamo andati a vedere cosa succedeva dentro la coclea di questi topi Em2/Em2. Già al primo giorno di vita (P1), lo spazio della Scala Media (quello che dovrebbe contenere l’endolinfa) era quasi inesistente, segno che la produzione di endolinfa “primaria” era compromessa fin da subito. Poi, crescendo (a P12 e 4 settimane), lo spazio si formava, ma la stria vascolare (StV) appariva più piccola, più spessa e come “accartocciata”, soprattutto nella parte basale della coclea. Le cellule della StV erano anche più “dense”, come se fossero più piccole e stipate.
Abbiamo usato una tecnica avanzata, la nano-tomografia computerizzata (nano-CT), per vedere meglio la struttura tridimensionale senza danneggiarla. Le immagini hanno confermato: la StV nei topi Em2/Em2 era significativamente più corta e ridotta in dimensioni.
Ma la scoperta forse più cruciale è arrivata dall’immunostaining, una tecnica che usa anticorpi fluorescenti per “vedere” dove si trova una specifica proteina. Nei topi normali (o eterozigoti Em2/+), la proteina Slc12a2 (NKCC1) si trova sulla membrana basolaterale delle cellule marginali della StV, il lato “giusto” per secernere ioni verso l’interno della StV. Nei topi Em2/Em2, invece, la proteina mutata (Slc12a2Δex21) era intensamente presente sulla membrana apicale, il lato sbagliato, quello rivolto verso l’endolinfa! Questo suggerisce che l’assenza dell’esone 21 causa un errore di “indirizzamento” (mistrafficking) della proteina.
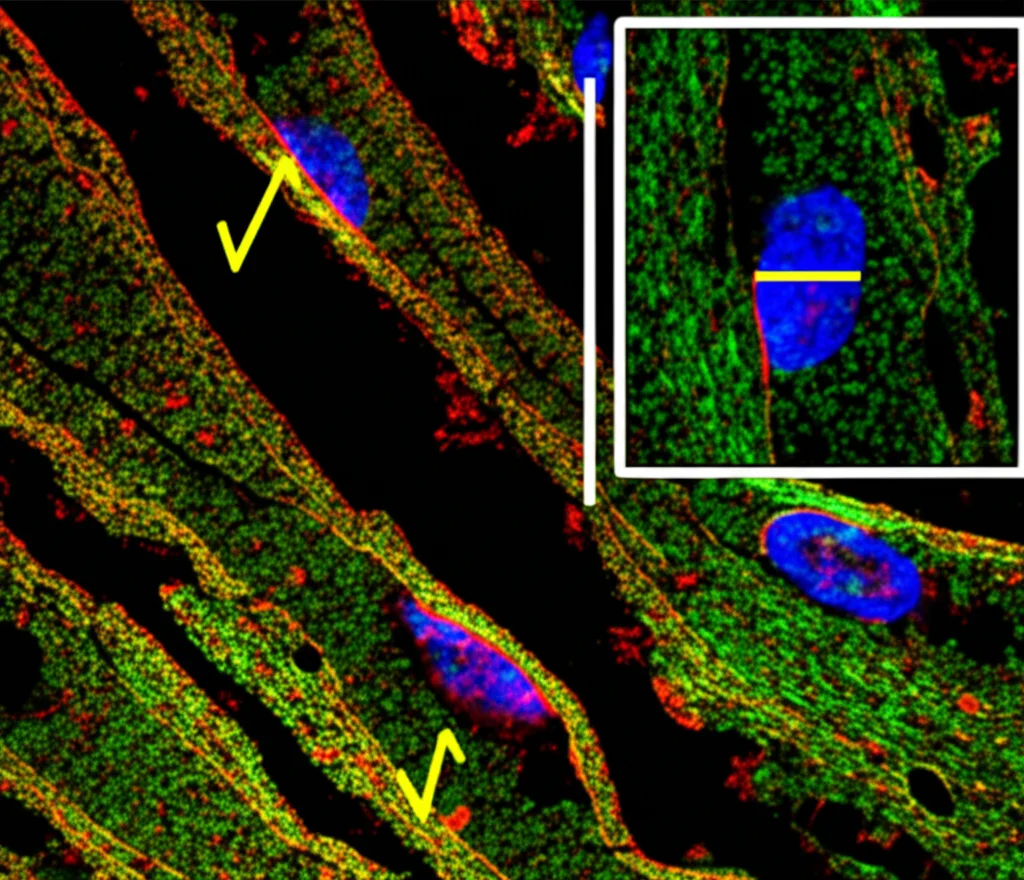
Indizi dal Genoma: l’RNA-seq Rivela una Risposta Inattesa
Per capire ancora meglio cosa succedeva a livello molecolare, abbiamo analizzato l’espressione di tutti i geni nella coclea dei topi Em2/Em2 usando l’RNA-seq. Abbiamo trovato centinaia di geni la cui attività era alterata rispetto ai topi normali. Molti geni sovra-regolati (cioè più attivi) erano legati all’adesione cellulare, in particolare alla formazione delle “giunzioni strette”, quelle che sigillano gli spazi tra le cellule.
Il gene più clamorosamente sovra-regolato era Cldn9, che codifica per la Claudina-9, una proteina delle giunzioni strette. L’aumento di Cldn9 era evidente a 4 settimane, ma non prima. Abbiamo ipotizzato un meccanismo affascinante: la mancanza di attività di trasporto di Slc12a2Δex21 e l’aumento di ioni nella StV (necessari per l’endolinfa “secondaria” che si forma dopo la nascita) potrebbero causare uno squilibrio osmotico, facendo “restringere” le cellule. Questo creerebbe delle “fessure” tra le cellule, compromettendo la barriera che isola l’endolinfa. L’aumento di Cldn9 potrebbe essere una risposta riparativa del tessuto, un tentativo di “tappare i buchi” e ripristinare la barriera, soprattutto nell’epitelio uditivo. Abbiamo infatti visto segnali più intensi di Cldn9 proprio nell’organo del Corti dei topi Em2/Em2.
Interessante notare che anche altri geni importanti per l’orecchio interno (come Gata3, Slc26a4, Tmprss3) erano sovra-regolati, forse un altro tentativo della coclea di compensare il difetto. Al contrario, geni legati alla segnalazione cellulare, al sistema nervoso e al trasporto ionico erano sotto-regolati.
La Sorpresa del Topo Em1: Non Tutti i Difetti Sono Uguali
E il topo Em1? Quello con la mutazione che credevamo equivalente a quella umana? Beh, qui abbiamo avuto una sorpresa! Analizzando l’RNA della coclea dei topi Em1/Em1, abbiamo trovato non solo il trascritto senza esone 21 (come in Em2), ma anche un trascritto più lungo. Sequenziandolo, abbiamo scoperto che questo trascritto più lungo conteneva sì l’esone 21, ma gli mancavano le prime 9 basi! Questo “splicing criptico” produceva una proteina Slc12a2 leggermente più corta (mancavano 3 amminoacidi), ma evidentemente ancora abbastanza funzionale. Risultato? I topi Slc12a2Em1/Em1 avevano un udito normale e una coclea morfologicamente normale!

Il Dettaglio che Fa la Differenza: Uomo vs Topo
Come mai la stessa (o quasi) mutazione nel sito di splicing dava risultati così diversi tra Em1 ed Em2, e perché Em1 non replicava la sordità umana? La risposta è arrivata da un esperimento chiamato “minigene assay” e da un confronto tra le sequenze del gene umano e murino. Abbiamo scoperto che c’è una singola differenza nucleotidica tra uomo e topo proprio all’inizio dell’esone 21 (una Timina nell’uomo, un’Adenina nel topo). Questo piccolo dettaglio cambia le regole dello splicing! Nel topo, la mutazione Em1 permette ancora l’uso di un sito di splicing alternativo (“criptico”) che salva gran parte dell’esone. Nell’uomo (o simulando la sequenza umana nel minigene), la stessa mutazione porta invece alla completa esclusione dell’esone 21. Ecco perché la mutazione Em1 nel topo non era un buon modello per la DFNA78 umana, mentre la Em2, che causa sempre l’omissione dell’esone, lo è!
Conclusioni e Prospettive Future
Insomma, questa ricerca ci ha insegnato tantissimo! Il topo Slc12a2Em2 si è rivelato un ottimo modello animale per studiare la patologia della sordità DFNA78, mostrando la sordità profonda, i problemi vestibolari e l’omissione dell’esone 21 proprio come nei pazienti. Abbiamo capito che l’assenza della regione codificata dall’esone 21 non solo blocca l’attività di trasporto ionico di NKCC1, ma causa anche un suo errato posizionamento nella cellula e scatena una serie di risposte compensatorie (come l’aumento di Cldn9) nel tentativo, purtroppo vano, di mantenere la funzione uditiva.
Certo, ci sono ancora tante domande aperte. Dobbiamo capire meglio perché le femmine Em2/+ mostrano un lieve deficit uditivo, misurare precisamente la composizione ionica dell’endolinfa e il potenziale elettrico nella coclea di questi topi, e identificare le molecole che interagiscono con la proteina normale e quella mutata. Ma ogni passo avanti ci avvicina a comprendere i meccanismi fini della sordità ereditaria e, speriamo, a trovare in futuro nuove strategie terapeutiche. È un lavoro complesso, ma incredibilmente stimolante!
Fonte: Springer