Caccia al Nuovo Farmaco Antimalarico: La Nostra Indagine Digitale su PfDHODH
Ciao a tutti! Oggi voglio portarvi con me in un viaggio affascinante nel mondo della ricerca farmaceutica, un’avventura combattuta non solo in laboratorio, ma anche grazie alla potenza dei computer. Stiamo parlando della lotta contro un nemico subdolo e persistente: la malaria.
La Malaria: Una Sfida Globale e la Corsa a Nuovi Farmaci
Come forse saprete, la malaria continua a mietere vittime, specialmente nelle regioni più povere del mondo. Pensate che nel 2023 si sono stimati circa 263 milioni di casi in 83 paesi, con un aumento di 11 milioni rispetto all’anno precedente! Il colpevole principale della forma più letale è un parassita chiamato Plasmodium falciparum. La cosa preoccupante è che questo parassita sta diventando sempre più resistente ai farmaci che abbiamo a disposizione, come le terapie combinate a base di artemisinina (ACT), che sono attualmente il trattamento di prima linea. Anche se abbiamo dei vaccini (RTS,S e R21), la necessità di trovare nuove armi terapeutiche è più urgente che mai.
Il Nostro Bersaglio: PfDHODH, un Enzima Chiave
Nella nostra ricerca, abbiamo puntato i riflettori su un bersaglio molto specifico all’interno del parassita: un enzima chiamato Diidroorotato Deidrogenasi di Plasmodium falciparum, o più semplicemente PfDHODH. Perché proprio lui? Beh, questo enzima è assolutamente fondamentale per la sopravvivenza del parassita. È coinvolto in un processo chiamato “sintesi delle pirimidine”, essenziale per costruire il DNA e permettere al parassita di replicarsi nel nostro corpo. La cosa fantastica è che l’enzima PfDHODH del parassita è strutturalmente diverso da quello umano. Questo ci dà una finestra di opportunità unica: possiamo progettare farmaci che blocchino specificamente l’enzima del parassita, senza (o con minimi) effetti collaterali su di noi. Bloccare PfDHODH significa impedire al parassita di produrre le “materie prime” per il suo DNA, fermando la sua divisione cellulare e portandolo alla morte. È come togliere un ingranaggio fondamentale da una macchina complessa.
I Protagonisti: Derivati Benzotiazinici
Il nostro studio si concentra su una classe specifica di molecole: i derivati della 3,4-Diidro-2H,6H-pirimido[1,2-c][1,3]benzotiazina-6-immina. Un nome un po’ lungo, lo so, ma queste molecole hanno mostrato un potenziale interessante come inibitori di PfDHODH. Abbiamo lavorato su un set di 43 di questi composti, già noti per la loro attività inibitoria.
Gli Strumenti del Mestiere: QSAR, Docking, Dinamica e Farmacocinetica
Qui entra in gioco la potenza della chimica computazionale. Per capire come queste molecole funzionano e come migliorarle, abbiamo usato un arsenale di tecniche digitali:
- QSAR (Quantitative Structure-Activity Relationship): Immaginatelo come un modo per trovare una “formula matematica” che collega la struttura chimica di una molecola alla sua efficacia contro il parassita. Analizzando i 43 composti, cerchiamo di capire quali “pezzi” della molecola sono più importanti per l’attività antimalarica.
- Docking Molecolare: È come simulare al computer l’incastro tra la nostra molecola (la chiave) e l’enzima PfDHODH (la serratura). Ci permette di vedere come la molecola si lega al sito attivo dell’enzima e quali interazioni chimiche la tengono ferma lì, bloccandone la funzione.
- Dinamica Molecolare (MD): Questa tecnica va oltre la semplice “fotografia” del docking. È come girare un filmato molecolare! Simuliamo il movimento degli atomi nel tempo per vedere quanto è stabile il legame tra la molecola e l’enzima e come entrambi si adattano l’uno all’altro.
- Studi di Farmacocinetica (ADME): Anche la molecola più potente è inutile se non riesce a raggiungere il bersaglio nel corpo o se viene eliminata troppo in fretta. Con questi studi computazionali (spesso chiamati in silico), prevediamo come la molecola verrà assorbita, distribuita, metabolizzata ed escreta dal corpo umano (ADME). Valutiamo anche la sua “drug-likeness”, cioè se ha le caratteristiche giuste per diventare un farmaco.

QSAR: Decifrare il Codice Struttura-Attività
Partendo dai nostri 43 composti e dai loro dati di attività (espressi come IC50, la concentrazione necessaria per inibire il 50% dell’enzima), abbiamo utilizzato software specifici (come ChemDraw, PaDEL-Descriptor e QSARINS) per costruire le strutture 3D, calcolare centinaia di descrittori molecolari (proprietà numeriche che descrivono la molecola) e sviluppare il modello QSAR. Abbiamo diviso i dati: il 75% per “allenare” il modello (training set) e il 25% per testarne l’affidabilità (test set).
Il risultato? Abbiamo ottenuto un modello QSAR molto robusto, basato su sette descrittori molecolari, con un coefficiente di determinazione (R²) pari a 0.92! Questo significa che il nostro modello è in grado di spiegare il 92% della variabilità nell’attività biologica osservata, un risultato eccellente. Anche altri parametri statistici ((Q^{2}) = 0.90, F = 43.57) confermano la bontà e la capacità predittiva del modello.
Ma cosa ci dice questo modello sulla relazione struttura-attività (SAR)? Abbiamo scoperto cose interessanti:
- Il numero di atomi di azoto (nN) sembra avere un impatto negativo: più azoto c’è, minore è l’attività.
- Descrittori legati alla superficie molecolare e alla idrofobicità (AATS5v) suggeriscono che un’eccessiva grandezza o idrofobicità può ridurre l’attività.
- La polarizzabilità degli atomi (AATS7p) è importante: atomi più polarizzabili favoriscono l’attività, suggerendo l’importanza delle interazioni elettroniche.
- L’ingombro sterico (MATS6c) gioca un ruolo: gruppi troppo voluminosi possono ostacolare il legame.
- La distribuzione elettronica (VR1_Dze, VR1_Dzs) è cruciale: la presenza e la posizione di atomi donatori o accettori di elettroni influenza significativamente l’attività.
- La forma complessiva della molecola (toposhape) è rilevante: certe forme si adattano meglio al sito attivo dell’enzima.
Abbiamo anche verificato il “Dominio di Applicabilità” (AD) del modello, per essere sicuri di fare previsioni affidabili solo per molecole simili a quelle usate per costruirlo. Solo 3 composti su 43 sono risultati leggermente fuori da questo dominio, ma con errori di previsione comunque accettabili.
Docking Molecolare: Trovare l’Incastro Perfetto
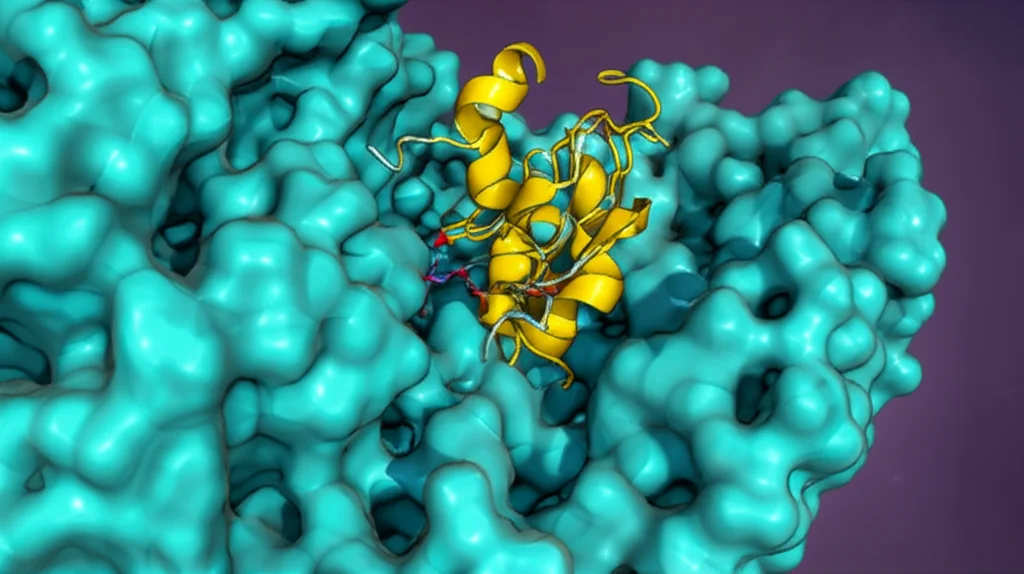
Successivamente, abbiamo usato il docking molecolare (con il software Glide di Schrödinger) per vedere come i nostri 43 composti si legano alla struttura 3D dell’enzima PfDHODH (abbiamo usato la struttura PDB ID: 6I4B). Abbiamo preparato sia le molecole che la proteina e poi abbiamo simulato l’aggancio nel sito attivo, dove normalmente si lega il suo substrato naturale (o altri inibitori noti).
I risultati sono stati molto promettenti! Molti dei nostri composti hanno mostrato affinità di legame (espresse come docking score in kcal/mol) migliori rispetto a farmaci di riferimento come Brequinar (-9.689 kcal/mol) e Artemisinina. Il nostro “campione” è stato il Composto 31, con un docking score eccezionale di -10.035 kcal/mol. Anche il Composto 01 (-9.677 kcal/mol), il Composto 14 (-9.62 kcal/mol) e il Composto 39 (-9.297 kcal/mol) si sono distinti.
Analizzando le interazioni, abbiamo visto che queste molecole si posizionano bene nella tasca idrofobica del sito attivo, vicino a dove si lega l’ubichinone (un cofattore dell’enzima). Formano legami idrogeno cruciali con residui aminoacidici chiave come HIS185 e interagiscono (interazioni steriche/idrofobiche) con altri come PHE171, ILE263, LEU176, CYS175. Ad esempio, nel Composto 31, l’anello pirimidinico forma un legame idrogeno con HIS185, mentre la presenza di un anello biciclico fuso sembra migliorare l’affinità. Queste interazioni sono simili a quelle osservate per altre classi note di inibitori di PfDHODH, come le triazolopirimidine e le chinoline, confermando che siamo sulla strada giusta.

Dinamica Molecolare: Osservare la Danza nel Tempo
Per essere ancora più sicuri, abbiamo preso i due composti più promettenti dal docking (Composto 31 e Composto 01) e li abbiamo sottoposti a simulazioni di dinamica molecolare (MD) per 50 nanosecondi usando GROMACS. Questo ci permette di vedere se il legame che abbiamo osservato nel docking è stabile nel tempo o se la molecola “scappa via”.
I risultati della MD sono stati confortanti. Abbiamo monitorato l’RMSD (Root Mean Square Deviation), una misura di quanto la posizione degli atomi cambia nel tempo rispetto alla posizione iniziale. Per entrambi i complessi (PfDHODH-Composto 31 e PfDHODH-Composto 01), l’RMSD si è stabilizzato rapidamente su valori bassi (circa 0.28-0.30 nm), fluttuando poi leggermente attorno a questi valori per tutta la simulazione. Questo indica che entrambi i composti formano un complesso stabile con l’enzima PfDHODH, rimanendo saldamente legati nel sito attivo. Il Composto 01 ha mostrato una flessibilità leggermente maggiore, ma entrambi i legami sono risultati robusti.
Farmacocinetica In Silico: Saranno Buoni Farmaci?
Infine, abbiamo valutato le proprietà farmacocinetiche e di “drug-likeness” dei 20 composti migliori emersi dal docking, usando il modulo QikProp di Schrödinger. Volevamo capire se queste molecole hanno le carte in regola per diventare potenzialmente dei farmaci somministrabili per via orale.
Abbiamo analizzato parametri chiave basati su regole farmacologiche consolidate:
- Regola dei Cinque di Lipinski: Valuta se la molecola ha dimensioni (peso molecolare < 500 Da), lipofilia (LogP < 5), e capacità di formare legami idrogeno adatte per l'assorbimento orale. La maggior parte dei nostri composti rispetta questa regola, con solo poche violazioni marginali (accettabili, ≤ 1 violazione) per composti come 31, 01, 18, 15, principalmente a causa di un LogP leggermente superiore.
- Regola di Veber: Considera l’area superficiale polare (PSA < 140 Ų) e il numero di legami ruotabili (< 10) per predire la biodisponibilità orale. I nostri composti rispettano anche questa regola.
- Regola di Egan: Predice la permeabilità passiva attraverso le membrane (LogP ≤ 5.88, PSA ≤ 131 Ų). Anche qui, la maggior parte dei composti rientra nei limiti.
- Assorbimento Orale Umano (% HOA): Tutti i composti selezionati hanno mostrato un eccellente assorbimento orale previsto, con valori superiori all’84%. Addirittura, i composti 31, 01, 04, 18 e 15 hanno raggiunto un teorico 100% di assorbimento orale!
- Permeabilità Caco-2 e MDCK: Queste simulano la capacità di attraversare le cellule intestinali. La maggior parte dei composti ha mostrato permeabilità da moderata ad alta, suggerendo un buon passaggio attraverso l’intestino. Il composto 16 è risultato meno permeabile, il che potrebbe richiedere aggiustamenti futuri.
- Penetrazione della Barriera Emato-Encefalica (QPlogBB): Idealmente, un farmaco antimalarico non dovrebbe entrare massicciamente nel cervello per evitare effetti collaterali sul sistema nervoso centrale. La maggior parte dei nostri composti ha mostrato valori bassi (< 0), indicando una scarsa penetrazione della BBB, il che è positivo. I composti 31 e 32 hanno valori leggermente più alti, un aspetto da tenere in considerazione.
- Permeabilità Cutanea (QPlogKp): Valutata per un’eventuale somministrazione transdermica, la maggior parte rientra nei range accettabili.
Abbiamo anche esaminato altri descrittori come il momento di dipolo, l’area superficiale accessibile al solvente (SASA), e il numero di donatori/accettori di legami idrogeno (HBD/HBA), che confermano ulteriormente il buon profilo generale di queste molecole.

Conclusioni e Prospettive Future
Quindi, cosa abbiamo imparato da questo viaggio computazionale? Abbiamo dimostrato che combinando QSAR, docking molecolare, dinamica molecolare e studi farmacocinetici in silico, possiamo ottenere informazioni preziose per la progettazione razionale di nuovi farmaci antimalarici.
Abbiamo identificato le caratteristiche strutturali chiave che rendono i derivati della 3,4-Diidro-2H,6H-pirimido[1,2-c][1,3]benzotiazina-6-immina attivi contro PfDHODH. Abbiamo individuato composti specifici, come il 31 e lo 01, che mostrano un’eccellente affinità di legame, interazioni favorevoli con il sito attivo e una stabilità confermata dalle simulazioni dinamiche. Inoltre, molti di questi composti presentano profili farmacocinetici promettenti, suggerendo che potrebbero essere sviluppati come farmaci orali efficaci.
Questo studio apre nuove strade per lo sviluppo di agenti antimalarici potenti, potenzialmente in grado di superare i problemi di resistenza che affliggono le terapie attuali. Certo, la strada è ancora lunga: questi risultati computazionali dovranno essere validati sperimentalmente in vitro e in vivo. Ma l’approccio multidimensionale che abbiamo utilizzato ci fornisce una solida base di partenza e una grande speranza nella lotta contro la malaria. È la dimostrazione di come la scienza computazionale possa essere un’alleata potente nella scoperta di nuovi farmaci per salvare vite umane.
Fonte: Springer