DNA Nascosto: Come i Mini-Domini nel Nucleo Controllano i Nostri Geni (Svelato da un Nuovo Modello!)
Ciao a tutti! Oggi voglio portarvi in un viaggio affascinante nel cuore delle nostre cellule, nel nucleo, dove si trova il manuale di istruzioni della vita: il DNA. Ma il DNA non è semplicemente un lungo filamento buttato lì a caso. Immaginate di dover stipare chilometri di filo in una scatola minuscola: avreste bisogno di organizzarlo per bene, no? Ecco, la cellula fa proprio questo con il DNA, impacchettandolo in una struttura complessa chiamata cromatina.
Questa organizzazione non è solo una questione di spazio, ma è fondamentale per decidere quali geni “accendere” e quali “spegnere”, influenzando tutto: come cresciamo, come funzionano i nostri organi, persino come ci ammaliamo.
Il Mistero dei Domini Nascosti
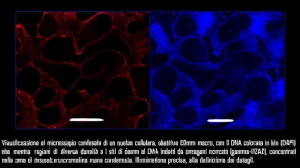
Da tempo sappiamo che la cromatina si divide grossolanamente in due tipi: l’eucromatina (zona A), più “aperta” e piena di geni attivi, e l’eterocromatina (zona B), super compatta e con geni per lo più silenziati. Ma grazie a tecniche di imaging potentissime, come la microscopia a super-risoluzione (tipo STORM), abbiamo scoperto che l’eterocromatina non è un blocco unico, ma è organizzata in tanti piccoli “domini” su scala nanometrica (parliamo di miliardesimi di metro!).
Questi mini-domini sono ovunque, in tantissimi tipi di cellule, ma come si formano esattamente? E soprattutto, come influenzano l’accensione e lo spegnimento dei geni? Sappiamo che la loro dimensione cambia se modifichiamo l’ambiente della cellula (ad esempio, la “rigidità” del materiale su cui cresce) o se usiamo farmaci che agiscono sull’epigenetica (quelle modifiche chimiche che decorano la cromatina, come l’acetilazione e la metilazione degli istoni, le proteine attorno cui si avvolge il DNA). Ma il meccanismo preciso era un bel rompicapo.
Un Modello per Svelare i Segreti del DNA
Ecco dove entra in gioco il nostro asso nella manica: abbiamo sviluppato un modello polimerico al computer che ci permette di simulare come si comporta la cromatina nel tempo e nello spazio. La cosa forte è che questo modello non parte da zero, ma integra dati reali provenienti da esperimenti di sequenziamento (come Hi-C, che mappa i contatti tra diverse parti del genoma, o ChIP-seq, che identifica le modifiche epigenetiche).
Immaginate il nostro modello come un filo di perline (la cromatina), dove ogni perlina rappresenta un pezzetto di genoma (circa 10.000 basi). Alcune perline sono “blu” (eucromatina, attiva) e altre “rosse” (eterocromatina, repressa), in base ai dati sperimentali. Queste perline interagiscono tra loro: quelle rosse si attraggono fortemente, formando grumi compatti, mentre le interazioni tra blu e rosse sono meno “amichevoli”.
Ma il bello viene ora: il modello non è statico! Le perline si muovono (simulando la diffusione nel nucleo) e, soprattutto, possono cambiare colore! Abbiamo introdotto delle “reazioni epigenetiche” che mimano l’azione degli enzimi cellulari (come HDAC, HAT, HMT, HDM) che aggiungono o rimuovono le etichette chimiche (acetilazione e metilazione) sugli istoni. Queste reazioni possono trasformare una perlina rossa in blu (acetilazione) e viceversa (metilazione), e la loro velocità può essere influenzata da segnali esterni, proprio come accade nelle cellule vere!

La Danza tra Diffusione e Reazioni: Nascono i Domini!
Facendo girare le simulazioni, abbiamo osservato qualcosa di straordinario. Se lasciamo agire solo la diffusione (il movimento e l’attrazione tra perline simili), le perline rosse tendono a formare un unico, enorme ammasso, separandosi completamente dalle blu. Questo fenomeno, noto come “maturazione di Ostwald”, non corrisponde a ciò che vediamo nelle cellule, dove ci sono tanti piccoli domini.
Ma quando attiviamo anche le reazioni epigenetiche, la musica cambia! Le reazioni (in particolare l’acetilazione, che trasforma rosso in blu) contrastano la tendenza all’aggregazione infinita. Si crea una sorta di “tiro alla fune” tra la diffusione, che vuole far crescere i domini rossi, e le reazioni, che li “smantellano” trasformando le perline rosse in blu.
Il risultato? Si formano domini di eterocromatina di dimensioni definite e stabili nel tempo, molto simili a quelli osservati al microscopio! E non solo: il nostro modello prevede una relazione matematica precisa (una “legge di scala”) tra la dimensione media di questi domini e la velocità delle reazioni epigenetiche. In pratica, più è veloce l’acetilazione (({Gamma }_{{ac}})), più piccoli diventano i domini ((R_d propto sqrt{D/{Gamma }_{{ac}}}), dove D è la diffusione). Allo stesso modo, la dimensione dipende dal rapporto tra metilazione (({Gamma }_{{me}})) e acetilazione ((R_d propto sqrt{{Gamma }_{{me}}/{Gamma }_{{ac}}({Gamma }_{{me}}+{Gamma }_{{ac}})})). È una scoperta fondamentale: abbiamo capito il meccanismo fisico che regola la taglia di queste strutture chiave!
Conferme Sperimentali: Melanoma sotto la Lente
Bello il modello, ma funziona nel mondo reale? Per verificarlo, abbiamo preso delle cellule di melanoma umano (A375) e le abbiamo trattate con un farmaco chiamato Tricostatina A (TSA). Il TSA è un inibitore delle HDAC, enzimi che rimuovono l’acetilazione. Quindi, trattare le cellule con TSA aumenta l’acetilazione globale, mimando un aumento della reazione ({Gamma }_{{ac}}) nel nostro modello.
Cosa ci aspettavamo? Domini di eterocromatina più piccoli. E cosa abbiamo visto con l’imaging STORM? Esattamente questo! I domini nelle cellule trattate con TSA erano significativamente più piccoli rispetto alle cellule di controllo, e la riduzione percentuale era incredibilmente simile a quella predetta dalle nostre simulazioni avviate con i dati specifici del melanoma. Bingo!
Ma non ci siamo fermati all’imaging. Abbiamo usato anche la tecnica Hi-C per mappare i contatti genomici su larga scala. Qui le cose si fanno interessanti. A livello “macro” (risoluzione di decine di migliaia di basi), i cambiamenti dopo il trattamento con TSA sembravano minimi. La maggior parte del genoma restava nel suo compartimento A o B. Sembrerebbe una contraddizione con l’imaging, ma il nostro modello ci aiuta a capire.

Il Segreto è nei Confini
Il modello, infatti, prevedeva un altro dettaglio cruciale: i cambiamenti epigenetici (da rosso a blu o viceversa) non avvengono a caso, ma si concentrano prevalentemente ai confini dei domini di eterocromatina. Perché? È una questione di energia: è più “facile” cambiare colore a una perlina al confine, dove le interazioni “amichevoli” (rosso-rosso) sono meno numerose, piuttosto che nel cuore compatto di un dominio rosso.
Ebbene, analizzando i dati Hi-C con più attenzione, abbiamo scoperto che quei pochi cambiamenti di compartimento osservati avvenivano proprio in prossimità dei confini tra le regioni A e B! Oltre il 75% dei “salti” da B ad A (da represso ad attivo, come ci si aspetta con TSA) si verificava entro 50.000 basi da un confine. Il modello aveva previsto correttamente anche questo!
Questa scoperta è potentissima: i confini dei domini sono le zone “calde”, i punti più sensibili dove l’ambiente esterno e i farmaci possono rimodellare la cromatina.
Dai Confini all’Espressione Genica (e alla Metastasi!)
Se i confini cambiano, cosa succede ai geni che si trovano lì vicino? Abbiamo analizzato l’espressione genica (con RNA-seq) nelle stesse cellule di melanoma trattate con TSA. Come previsto, c’era una tendenza generale all’aumento dell’espressione genica (più geni accesi). Ma la cosa più intrigante è che i geni situati vicino ai confini che passavano da B ad A mostravano un aumento significativo dell’espressione.
E quali geni si trovavano in queste zone “sensibili”? Analizzando le loro funzioni, abbiamo trovato un arricchimento significativo di geni coinvolti nella migrazione cellulare e in percorsi chiave come WNT e MAPK/ERK, noti per essere cruciali nella transizione epitelio-mesenchimale (EMT). L’EMT è un processo fondamentale per la capacità delle cellule tumorali di staccarsi, muoversi e formare metastasi. Quindi, il nostro modello suggerisce un meccanismo diretto: i cambiamenti ambientali o farmacologici modificano l’epigenetica ai confini dei domini, questo porta alla de-compattazione locale della cromatina e all’attivazione di geni cruciali per la progressione tumorale, come quelli dell’EMT.
Non Solo Melanoma: Conferme nelle Cellule Staminali
Per dimostrare che non si trattava di un caso isolato, abbiamo applicato il nostro modello a un altro sistema: le cellule staminali mesenchimali umane (hMSC). È noto che queste cellule cambiano l’organizzazione della loro cromatina in risposta alla rigidità del substrato su cui crescono: su substrati più rigidi, la cromatina è più “aperta” (meno eterocromatina compatta), mentre su quelli più morbidi è più compatta. Questo avviene perché la rigidità influenza la quantità di certi enzimi epigenetici (come EZH2, una metiltransferasi) nel nucleo.
Abbiamo simulato questo scenario, usando dati ChIP-seq delle hMSC per inizializzare il modello e variando i tassi di reazione per mimare l’effetto della rigidità (più rigidità = meno metilazione). Le simulazioni hanno predetto una diminuzione della dimensione dei domini di eterocromatina su substrati più rigidi. Abbiamo confrontato queste predizioni con dati sperimentali di STORM imaging pubblicati in precedenza: la corrispondenza era eccellente!
Abbiamo anche simulato l’effetto di un inibitore specifico di EZH2 (GSK343, o GSK). Anche in questo caso, il modello ha predetto correttamente la riduzione delle dimensioni dei domini osservata sperimentalmente con STORM. Questo conferma la versatilità del nostro approccio: lo stesso meccanismo (bilanciamento diffusione/reazioni ai confini) spiega l’organizzazione della cromatina in contesti diversi, dalla risposta ai farmaci alla meccano-sensibilità.

I Confini Custodiscono la Memoria della Cellula
C’è un’ultima implicazione affascinante. Le cellule spesso “ricordano” gli ambienti che hanno incontrato. Questa memoria epigenetica è cruciale, ad esempio, durante lo sviluppo o in malattie come il cancro. Come viene immagazzinata questa memoria nella struttura della cromatina?
Abbiamo usato il nostro modello per simulare uno scenario di “memoria”: abbiamo esposto le hMSC simulate prima a un forte stimolo di acetilazione (come un ambiente rigido o un farmaco) per un certo tempo, e poi a uno stimolo opposto di metilazione (ambiente morbido o rimozione del farmaco).
Abbiamo osservato tre tipi di comportamento per le perline (i segmenti di cromatina):
- Conservate: Non cambiano mai colore, indipendentemente dagli stimoli. Rappresentano regioni genomiche stabili.
- Recuperate: Cambiano colore durante il primo stimolo (es. da rosso a blu) ma tornano al colore originale quando lo stimolo cessa. Sono regioni “elastiche”.
- Memoria dallo shock: Cambiano colore durante il primo stimolo e *non* tornano indietro, mantenendo il nuovo stato anche dopo il cambio di stimolo. Queste sono le regioni che portano la memoria!
Indovinate dove si trovavano prevalentemente le regioni “memoria” e “recuperate”? Esatto, ancora una volta, ai confini dei domini o all’interno di domini molto piccoli (che si comportano quasi interamente come confini). I grandi domini di eterocromatina tendono a rimanere “conservati” nel loro nucleo. Questo suggerisce che i confini non sono solo sensori del presente, ma anche registratori del passato della cellula! La proporzione di regioni “memoria” aumentava con l’intensità dello stimolo iniziale, indicando che esperienze più “forti” lasciano un segno più duraturo.

Conclusioni: Un Nuovo Livello di Comprensione (e Nuove Domande!)
Quindi, cosa abbiamo imparato da questo viaggio?
- Abbiamo sviluppato un modello potente che integra dati sperimentali e simulazioni per studiare l’organizzazione dinamica della cromatina.
- Abbiamo scoperto che la formazione e la dimensione dei domini nanoscopici di eterocromatina derivano da un equilibrio tra diffusione passiva e reazioni epigenetiche attive.
- Abbiamo identificato i confini di questi domini come i punti cruciali dove avvengono i cambiamenti epigenetici in risposta a segnali esterni (meccanici o chimici).
- Abbiamo dimostrato che questi cambiamenti ai confini influenzano direttamente l’espressione dei geni vicini, con potenziali conseguenze su processi come la metastasi tumorale.
- Abbiamo ipotizzato che i confini siano anche i siti dove si deposita la memoria epigenetica della cellula.
Questo lavoro apre nuove prospettive per capire come le cellule rispondono al loro ambiente a livello del genoma. Ci aiuta a comprendere meglio malattie complesse come il cancro e potrebbe, in futuro, suggerire nuove strategie terapeutiche mirate a rimodellare l’organizzazione della cromatina. Certo, il nostro modello è una semplificazione e ci sono ancora tanti dettagli da esplorare (altri tipi di modifiche epigenetiche, il ruolo di altre proteine, l’organizzazione tra cromosomi diversi…), ma crediamo sia un passo importante per decifrare il codice nascosto nell’architettura del nostro DNA. È un campo di ricerca in continua evoluzione, ed è entusiasmante essere parte di questa avventura!
Fonte: Springer