Sclerosi Multipla: Ho Trovato Come Spegnere l’Interruttore dell’Infiammazione nel Cervello!
Ciao a tutti! Oggi voglio parlarvi di qualcosa che mi appassiona tantissimo e che potrebbe aprire nuove strade per combattere malattie toste come la Sclerosi Multipla (SM). Immaginate il nostro sistema nervoso centrale (SNC) come una città super complessa e trafficata. A volte, però, alcune cellule del nostro stesso sistema immunitario, che dovrebbero difenderci, si confondono e iniziano ad attaccare parti della città, come la guaina mielinica che protegge i nostri “cavi” neuronali. Questo è quello che succede, in parole povere, nella SM e nel suo modello animale che usiamo per studiarla, l’encefalomielite autoimmune sperimentale (EAE).
I Protagonisti di Questa Battaglia
Due tipi di cellule sono i principali “colpevoli” in questo scenario: le microglia e le cellule Th17. Le microglia sono come le guardie residenti del nostro cervello, cellule immunitarie che vivono lì stabilmente. Le Th17, invece, sono un tipo specifico di linfociti T, soldati del sistema immunitario, che in queste malattie diventano particolarmente aggressivi contro il nostro stesso corpo (autoimmuni).
Sappiamo da tempo che entrambe giocano un ruolo chiave, ma una domanda mi frullava in testa: queste due cellule “parlano” tra loro all’interno del cervello infiammato? E se sì, come? La mia ipotesi era che le microglia, che possono “presentare” pezzi del nemico (antigeni) alle altre cellule immunitarie, potessero attivare e “caricare” le cellule Th17 patogene proprio lì, nel SNC. E non solo: pensavo che le Th17, una volta attivate, potessero a loro volta “dare la carica” alle microglia, creando un circolo vizioso, un loop di attivazione che auto-alimenta l’infiammazione. Un vero e proprio “feed-forward loop”.
La Caccia al Circolo Vizioso
Per vederci chiaro, abbiamo usato diversi modelli sperimentali su topolini, inclusi quelli con EAE indotta attivamente o passivamente (trasferendo cellule immunitarie già “arrabbiate”) e topolini geneticamente modificati. Ebbene sì, i dati ci hanno dato ragione! Abbiamo dimostrato che esiste proprio questo loop di attivazione microglia-Th17 che spinge avanti la malattia.
Come funziona questo dialogo infuocato? È un meccanismo complesso che dipende da più fattori:
- Le microglia devono esporre sulla loro superficie una molecola chiamata MHC-II per “presentare” l’antigene alle Th17.
- Le microglia rilasciano citochine pro-infiammatorie (messaggeri chimici che accendono l’infiammazione) e chemochine (che attirano altre cellule immunitarie sul posto).
- Un percorso molecolare specifico all’interno delle microglia, chiamato STING→NF-κB, sembra essere fondamentale per mantenere le microglia in questo stato “iperattivo”.
- Le cellule Th17 patogene, una volta attivate, rispondono producendo le loro citochine “da battaglia”, come GM-CSF e IFNγ, che a loro volta gettano benzina sul fuoco attivando ulteriormente le microglia.
Siamo riusciti persino a “fotografare” queste coppie di cellule (microglia-Th17) mentre interagivano, dimostrando che sono le unità funzionali dove avviene questa presentazione dell’antigene e l’attivazione reciproca. È come aver colto sul fatto i due complici mentre pianificano l’attacco!
Una Possibile Arma Segreta: ACT001
E qui arriva una parte ancora più emozionante. Esiste un farmaco, chiamato ACT001, che è già stato approvato come farmaco orfano negli USA per trattare un tipo di tumore al cervello, il glioblastoma. Questo farmaco deriva da un composto naturale (micheliolide) e ha delle proprietà molto interessanti: attraversa la barriera emato-encefalica (cioè arriva nel cervello), ha bassa tossicità e ha già mostrato di poter ridurre l’infiammazione cerebrale in altri contesti (come il trauma cranico).
Mi sono chiesto: e se ACT001 potesse funzionare anche nell’EAE, magari proprio interrompendo quel dannato circolo vizioso microglia-Th17? Abbiamo deciso di provare.
Quando Agisce ACT001? Il Tempismo è Tutto!
Per capire come e quando ACT001 potesse essere utile, abbiamo trattato i topolini con EAE in diversi momenti della malattia. L’EAE ha due fasi principali: una fase di induzione (quando il sistema immunitario viene “addestrato” ad attaccare la mielina, ma i sintomi non sono ancora evidenti) e una fase effettrice (quando le cellule immunitarie attaccano attivamente il SNC e compaiono i sintomi come la paralisi).
Abbiamo diviso i topi in gruppi: alcuni trattati con ACT001 fin dall’inizio (fase di induzione + effettrice), altri solo durante l’induzione, altri solo durante la fase effettrice, e un gruppo di controllo senza trattamento. I risultati sono stati chiarissimi:
- Trattare solo durante la fase di induzione? Nessun effetto. La malattia progrediva come se nulla fosse.
- Trattare durante entrambe le fasi o solo durante la fase effettrice? Bingo! La gravità dei sintomi si riduceva significativamente.
Analizzando il midollo spinale (uno dei bersagli principali dell’attacco autoimmune), abbiamo visto che nei topi trattati efficacemente con ACT001 (cioè durante la fase effettrice) c’era molta meno infiltrazione di cellule immunitarie e quasi nessuna demielinizzazione, rispetto ai gruppi non trattati o trattati solo all’inizio. Anche i livelli di citochine infiammatorie (come Il1b, Csf2, Il6, Il17a, Ifng) nelle cellule infiltrate erano molto più bassi, mentre aumentava una citochina anti-infiammatoria (Il10).
Questo ci ha detto una cosa fondamentale: ACT001 agisce proprio quando l’infiammazione è già in corso nel cervello, durante la fase effettrice. Ed è sufficiente trattare solo in questa fase per ottenere benefici.
Chi è il Vero Bersaglio di ACT001?
Ok, ACT001 funziona nella fase effettrice. Ma come? Quali cellule colpisce? Dato che le Th17 sono così importanti, abbiamo pensato fossero loro il bersaglio principale. Abbiamo analizzato le cellule T CD4+ (la famiglia a cui appartengono le Th17, ma anche le Th1 (altre cellule pro-infiammatorie) e le Treg (cellule regolatrici, che cercano di calmare l’infiammazione)) nei linfonodi e nel midollo spinale dei topi trattati e non.
Sorpresa! Nei linfonodi (dove avviene l’addestramento iniziale), ACT001 non cambiava quasi nulla: numero totale di cellule T CD4+, numero di Treg, Th1 o Th17… tutto normale. Ma nel midollo spinale (il campo di battaglia), le cose cambiavano eccome! Il numero totale di cellule T CD4+ diminuiva. Le Treg e le Th1 rimanevano più o meno stabili, ma le Th17, specialmente quelle più “cattive” che producono anche IFNγ (IFNγ+Th17) o GM-CSF (GM-CSF+ Th17), erano significativamente ridotte dal trattamento con ACT001. Anche l’espressione dei geni chiave per la funzione delle Th17 (come Il17a e Rorc) era più bassa nelle cellule T isolate dal midollo spinale dei topi trattati.
Questo sembrava confermare che ACT001 colpisse le Th17, ma solo nella fase di attivazione nel SNC, non durante il loro “addestramento” iniziale nei linfonodi. Per esserne sicuri, abbiamo fatto esperimenti di trasferimento adottivo: abbiamo preso cellule T CD4+ da topi donatori con EAE (già “addestrate”), le abbiamo espanse in laboratorio (simulando la fase di priming) e poi le abbiamo trasferite in topi riceventi sani, che a quel punto sviluppavano l’EAE (simulando la fase di attivazione nel SNC).
- Se trattavamo con ACT001 durante la fase di priming (nel donatore e in laboratorio)? Nessun effetto sulla capacità di queste cellule di scatenare la malattia nel ricevente.
- Se trattavamo con ACT001 solo il topo ricevente, durante la fase di attivazione nel SNC? Ecco che la malattia era molto più lieve, con meno infiammazione e meno Th17 attive nel midollo spinale.
Abbiamo anche provato a vedere se ACT001 influenzasse direttamente la differenziazione delle cellule T CD4+ “naive” (vergini) in Th17, Th1 o Treg in provetta. Risultato? Nessun effetto significativo.
Il Colpo di Scena: Non Sono le Th17, Sono le Microglia!
Qui le cose si facevano strane. ACT001 inibiva potentemente l’attivazione delle Th17 nel cervello in vivo, ma non sembrava avere effetto diretto su di loro in vitro o durante il priming. Come era possibile? L’unica spiegazione era che ACT001 non agisse direttamente sulle Th17, ma su qualcun altro che interagiva con loro nel SNC… e i sospetti ricadevano pesantemente sulle microglia!
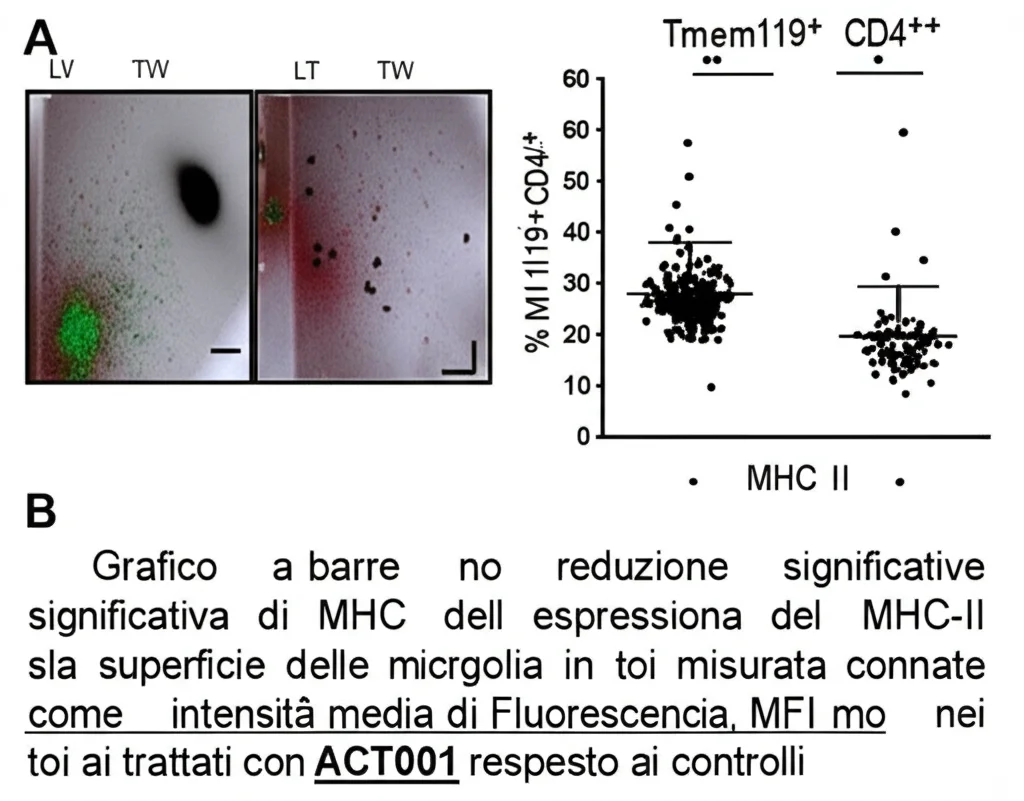
Siamo andati a vedere cosa succedeva alle microglia nei topi trattati con ACT001 durante la fase effettrice. Ed ecco la scoperta chiave: ACT001 cambiava lo “stato” delle microglia! Riduceva la proporzione di microglia “attive” (iper-infiammatorie, CD45HiCD11b+) e aumentava quelle “omeostatiche” (più calme, CD45dimCD11b+). Le microglia trattate esprimevano meno molecole associate all’attivazione infiammatoria come MHC-II e iNOS, e più molecole legate a uno stato di quiete, come CD206 (Mrc1). A livello di geni, ACT001 spegneva quelli pro-infiammatori (Nos2, Il1b, Tnf) e accendeva quelli calmanti (Mrc1, Il10).
Quindi, ACT001 riportava le microglia verso uno stato di normalità, meno “arrabbiato”. E la riduzione di MHC-II era particolarmente interessante, perché suggeriva che ACT001 potesse interferire proprio con la capacità delle microglia di presentare l’antigene e attivare le Th17!
Abbiamo usato una tecnica speciale (citometria a flusso) per “vedere” le interazioni fisiche tra microglia (marcate con Tmem119) e cellule T CD4+ nel midollo spinale. Siamo riusciti a identificare le cellule singole e le “coppie” (doublets) di microglia e T cellula che stavano interagendo. Indovinate un po’? Le microglia che interagivano con le cellule T esprimevano livelli molto più alti di MHC-II rispetto a quelle “single”. E ACT001 non solo riduceva l’espressione di MHC-II sulle microglia in generale, ma diminuiva anche il numero di queste interazioni microglia-T cellula!
L’Interruttore Molecolare: La Via STING→NF-κB
Per capire ancora più a fondo come ACT001 calmasse le microglia, abbiamo analizzato tutti i geni espressi dalle microglia isolate dal midollo spinale dei topi trattati e non (RNA-seq). ACT001 spegneva geni legati all’attivazione microgliale (Il1b, Il6, Tnf, Csf2) e alle vie infiammatorie come TNF, JAK-STAT e, soprattutto, la via NF-κB. Curiosamente, erano downregolate anche vie legate al riconoscimento di DNA nel citoplasma, come la via cGAS-STING.
La via cGAS-STING è un sistema di allarme cellulare che rileva DNA fuori posto (ad esempio da cellule danneggiate o da virus) e attiva una risposta infiammatoria, spesso proprio tramite NF-κB. Nel cervello, questa via può essere attivata nelle microglia da detriti cellulari o DNA mitocondriale “perso”, contribuendo a varie malattie neuroinfiammatorie, inclusa l’EAE/SM.
La nostra ipotesi è diventata: e se la via STING→NF-κB fosse iperattiva nelle microglia durante l’EAE, mantenendole accese e alimentando il loop con le Th17? E se ACT001 funzionasse proprio bloccando questo interruttore STING→NF-κB?
Abbiamo verificato: nelle microglia dei topi con EAE, STING e NF-κB (la proteina p65) erano effettivamente più “accesi” (fosforilati) e NF-κB si spostava nel nucleo per attivare i geni infiammatori. Questa attivazione aumentava con la progressione della malattia. E cosa faceva ACT001? Riduceva l’attivazione di STING e NF-κB nelle microglia dei topi malati!
Abbiamo confermato questo effetto in laboratorio, su microglia primarie. Stimolando le microglia con attivatori della via STING (come 2’3′-cGAMP o il virus HSV-1), queste si accendevano producendo citochine infiammatorie. ACT001 riusciva a bloccare l’attivazione di STING e NF-κB e a ridurre la produzione di queste citochine in modo dose-dipendente. È interessante notare che ACT001 era molto meno efficace nel bloccare l’attivazione indotta da LPS (un componente batterico che attiva NF-κB per altre vie), suggerendo che il suo bersaglio fosse proprio la via dipendente da STING.
La Prova del Nove: Topi Senza STING
Per essere sicuri al 100% che STING fosse il mediatore chiave dell’effetto di ACT001, abbiamo indotto l’EAE in topolini normali (WT) e in topolini geneticamente modificati a cui mancava il gene STING (Sting1-/-). Risultato? I topi senza STING sviluppavano una forma molto più lieve di EAE, con meno infiammazione e meno attivazione delle Th17. Questo già confermava l’importanza di STING nella malattia. Ma la cosa cruciale è stata che, trattando questi topi Sting1-/- con ACT001, il farmaco non dava alcun beneficio aggiuntivo! La malattia rimaneva lieve come nei topi Sting1-/- non trattati. Questa è stata la prova definitiva: l’effetto terapeutico di ACT001 nell’EAE passa proprio attraverso il blocco della via di STING.
 Springer
Springer





