PVT1: Il Regista Occulto che Accelera il Cancro al Fegato? La Nostra Indagine Rivela Nuovi Bersagli
Ciao a tutti! Oggi voglio portarvi nel cuore della ricerca sul cancro, in particolare su una forma molto aggressiva: il carcinoma epatocellulare (HCC), il tipo più comune di cancro al fegato. Sapete, questa malattia non solo mette a dura prova il fisico, ma anche la mente dei pazienti, e purtroppo le terapie attuali, sebbene in evoluzione, non sono ancora abbastanza efficaci per tutti e i bersagli molecolari che possiamo colpire sono limitati.
Il Mistero dell’HCC e l’Intrigante Ruolo di PVT1
L’HCC è un nemico subdolo. Si piazza al sesto posto tra tutti i tumori per incidenza e al terzo per mortalità a livello globale. Le cause principali le conosciamo: epatite B e C croniche, abuso di alcol… ma i meccanismi precisi che lo fanno nascere e crescere restano in parte avvolti nel mistero. Ed è qui che entriamo in gioco noi ricercatori, cercando di svelare questi meccanismi per trovare nuove armi.
Negli ultimi anni, l’attenzione si è concentrata su delle molecole affascinanti chiamate lncRNA (long non-coding RNA). Immaginatele come dei lunghi filamenti di RNA che, a differenza dei loro cugini più famosi (gli mRNA), non servono a costruire proteine, ma agiscono come dei sofisticati regolatori all’interno della cellula. Uno di questi, chiamato lncRNA PVT1 (Plasmacytoma Variant Translocation 1), è finito sotto i nostri riflettori. Diversi studi lo avevano già collegato alla progressione di vari tumori, incluso l’HCC, suggerendo che potesse accelerarne la crescita e la capacità di diffondersi (le temute metastasi), influenzando negativamente la sopravvivenza dei pazienti. Ma *come* esattamente? Il meccanismo preciso era sfuggente.
TGF-β1: Un Attore dal Doppio Volto e la Connessione con PVT1
Un altro protagonista noto nell’HCC è il TGF-β1. Questa molecola è un po’ un Giano Bifronte: da un lato può *inibire* la crescita delle cellule tumorali, ma dall’altro può *aumentare* la loro capacità di invadere altri tessuti e formare metastasi. Un bel paradosso, vero? Bloccare completamente il TGF-β1 non è una strada percorribile a lungo termine, perché è coinvolto anche in processi fisiologici normali e ci sarebbero troppi effetti collaterali. Ecco perché identificare i suoi “soldati” a valle, le molecole che mediano i suoi effetti pro-metastatici, diventa cruciale.
La nostra indagine è partita proprio da qui: potevamo svelare un nuovo meccanismo a valle del TGF-β1 nell’HCC e capire più a fondo come PVT1 facesse il suo “sporco lavoro”? L’obiettivo era ambizioso: fornire nuove prove e, potenzialmente, nuovi bersagli per terapie geniche mirate contro l’HCC.
Analizzando dati pubblici (come il database GEPIA e Kaplan-Meier Plotter) e conducendo esperimenti preliminari su diverse linee cellulari di HCC (HepG2, Hep3B, Huh7, PLC/PRF/5, MHCC97H, HCCLM3) e cellule epatiche normali (THLE-3), abbiamo avuto le prime conferme: PVT1 era effettivamente molto più espresso nelle cellule tumorali rispetto a quelle normali, e alti livelli di PVT1 erano associati a una prognosi peggiore per i pazienti. Ma la vera sorpresa è arrivata quando abbiamo trattato le cellule tumorali con TGF-β1: i livelli di PVT1 schizzavano alle stelle! E, viceversa, bloccando il TGF-β1 con un inibitore (SB431542), l’espressione di PVT1 diminuiva. Bingo! Avevamo trovato un legame diretto: il TGF-β1 è un regolatore a monte di PVT1.
Smad3: Il Messaggero che Attiva PVT1
Ma come fa il TGF-β1 a “parlare” con PVT1? Abbiamo pensato alla via di segnalazione classica del TGF-β, quella che coinvolge le proteine Smad. In questa via, il TGF-β1 attiva Smad3, che poi entra nel nucleo della cellula e agisce come un fattore di trascrizione, accendendo o spegnendo specifici geni. Poteva essere Smad3 il messaggero tra TGF-β1 e PVT1?
Per verificarlo, abbiamo “silenziato” Smad3 nelle cellule tumorali usando delle piccole molecole di RNA (siRNA). Il risultato? L’espressione di PVT1 è crollata. Questo ci ha detto che Smad3 era necessario per mantenere alti i livelli di PVT1. Ma Smad3 si legava direttamente al “promotore” di PVT1 (la regione di DNA che ne controlla l’accensione)? Con tecniche sofisticate come la ChIP (Chromatin Immunoprecipitation) e i saggi con geni reporter luciferasi, abbiamo dimostrato che sì, Smad3 si lega proprio al promotore di PVT1 e, quando attivato da TGF-β1, ne aumenta l’espressione. Era come aver trovato l’interruttore!
PVT1: Il Motore della Crescita e della Diffusione Tumorale
Ora che sapevamo *chi* accendeva PVT1, volevamo capire nel dettaglio *cosa* facesse PVT1 una volta acceso. Abbiamo creato modelli cellulari in cui potevamo spegnere PVT1 (knockdown con shRNA) o accenderlo ancora di più (sovraespressione).
I risultati sono stati netti:
- Spegnere PVT1: Le cellule tumorali rallentavano la loro divisione (si bloccavano in una fase specifica del ciclo cellulare, G0/G1), formavano meno colonie e, soprattutto, perdevano la loro capacità di migrare e invadere (come misurato con test di “wound healing” e Transwell).
- Accendere PVT1: Effetto opposto! Le cellule proliferavano più velocemente e diventavano più aggressive, più capaci di muoversi e invadere.
Era chiaro: PVT1 agisce come un acceleratore sia per la crescita che per la diffusione metastatica dell’HCC.
Svelare i Complici di PVT1: Smad6 e NRG1
Ma come fa PVT1 a orchestrare tutto questo? I lncRNA spesso funzionano come delle “spugne molecolari” (un meccanismo chiamato ceRNA, competitive endogenous RNA). Immaginate che PVT1 possa “assorbire” dei piccoli RNA regolatori chiamati microRNA (miRNA), impedendo loro di fare il loro lavoro, che di solito è quello di silenziare altri geni (mRNA).
Analizzando dati di sequenziamento RNA e usando strumenti bioinformatici, abbiamo identificato due potenziali “vittime” di questo meccanismo, due geni che sembravano essere regolati da PVT1:
- Smad6: Un membro della famiglia Smad, ma con un ruolo *inibitorio*, specialmente sulla via di segnalazione BMP (Bone Morphogenetic Protein), che a sua volta può attivare geni che bloccano il ciclo cellulare, come P21. Abbiamo scoperto che quando PVT1 era alto, anche Smad6 era alto, mentre P21 era basso. L’ipotesi? PVT1 potrebbe aumentare Smad6, che a sua volta inibisce la via BMP/P21, togliendo così un freno alla proliferazione cellulare.
- NRG1 (Neuregulin 1): Una molecola chiave nella trasformazione epitelio-mesenchimale (EMT), un processo fondamentale per l’invasione e la metastasi. NRG1 attiva recettori sulla superficie cellulare (come ERBB2/ERBB3) che scatenano segnali pro-migrazione. Anche qui, quando PVT1 era alto, NRG1 era alto. L’ipotesi? PVT1 potrebbe promuovere le metastasi aumentando NRG1.
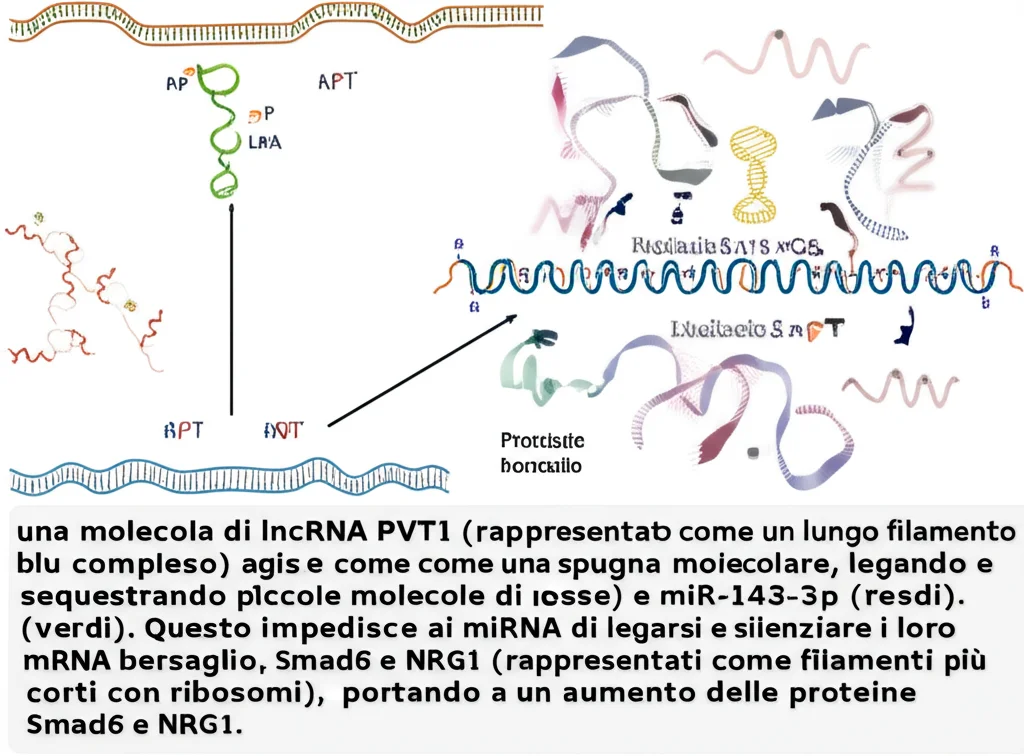
Le Spugne Molecolari: miR-186-5p e miR-143-3p
Ma quali erano i miRNA che PVT1 “sequestrava” per regolare Smad6 e NRG1? La nostra ricerca bioinformatica e la letteratura ci hanno messo sulla pista giusta:
- Per Smad6: Abbiamo identificato miR-186-5p. Era già noto che miR-186-5p potesse legare e inibire Smad6 e che PVT1 potesse interagire con miR-186-5p. Abbiamo confermato sperimentalmente: PVT1 lega miR-186-5p, impedendogli di silenziare Smad6. Risultato netto: più PVT1 = più Smad6 libero di agire.
- Per NRG1: Abbiamo puntato su miR-143-3p. Anche in questo caso, era noto che miR-143-3p potesse inibire NRG1 e che diversi lncRNA potessero “spugnare” miR-143-3p nell’HCC. I nostri esperimenti hanno confermato: PVT1 lega miR-143-3p, impedendogli di silenziare NRG1. Risultato netto: più PVT1 = più NRG1 libero di promuovere la metastasi.
Avevamo scoperto i due assi principali attraverso cui PVT1 agisce: l’asse PVT1/miR-186-5p/Smad6 per la proliferazione e l’asse PVT1/miR-143-3p/NRG1 per la metastasi.
La Prova del Nove: Gli Esperimenti di “Salvataggio”
Per essere assolutamente sicuri che fossero proprio questi gli ingranaggi del meccanismo, abbiamo fatto degli esperimenti chiamati “rescue” (salvataggio), sia in vitro (sulle cellule) che in vivo (su modelli animali, topolini nude).
L’idea è semplice: se spengo PVT1 e la cellula smette di proliferare, cosa succede se “restituisco” alla cellula solo Smad6 (il bersaglio a valle)? Se la proliferazione riparte, significa che PVT1 agisce proprio tramite Smad6. E viceversa: se accendo PVT1 e la cellula prolifera di più, cosa succede se spengo Smad6? Se la proliferazione rallenta, la conferma è doppia.
Abbiamo fatto proprio questo, giocando con diverse combinazioni:
- PVT1 spento + Smad6 riacceso: La proliferazione cellulare (e la crescita del tumore nei topi) veniva “salvata”, cioè riprendeva vigore. Ma la capacità di metastasi non cambiava.
- PVT1 acceso + Smad6 spento: La proliferazione accelerata da PVT1 veniva bloccata.
- PVT1 spento + NRG1 riacceso: La capacità di migrazione/invasione (e le metastasi polmonari nei topi) veniva “salvata”. Ma la proliferazione non cambiava.
- PVT1 acceso + NRG1 spento: La capacità metastatica aumentata da PVT1 veniva ridotta.
Abbiamo ripetuto esperimenti simili giocando anche con i livelli dei miRNA (usando inibitori o “mimics” per aumentarne o diminuirne l’effetto). I risultati hanno confermato tutto: PVT1 promuove la proliferazione sequestrando miR-186-5p per aumentare Smad6, e promuove la metastasi sequestrando miR-143-3p per aumentare NRG1.
Conclusioni: PVT1 Come Bersaglio Terapeutico Promettente
Questa ricerca ci ha permesso di dipingere un quadro molto più chiaro del ruolo di PVT1 nell’epatocarcinoma. Abbiamo svelato come sia attivato dalla via TGF-β1/Smad3 e come, a sua volta, agisca da regista molecolare promuovendo la crescita tumorale tramite l’asse miR-186-5p/Smad6 e la diffusione metastatica tramite l’asse miR-143-3p/NRG1.
Perché è importante? Perché ci offre un nuovo potenziale bersaglio terapeutico. Colpire direttamente TGF-β1 è complicato, ma colpire PVT1 potrebbe essere una strategia più mirata e forse più sicura per contrastare sia la proliferazione che le metastasi dell’HCC. Certo, la strada verso una terapia è ancora lunga, ma aver identificato un attore così centrale e averne svelato i meccanismi d’azione è un passo avanti fondamentale. Speriamo che queste scoperte aprano la via a nuove strategie di trattamento per i pazienti affetti da questa difficile malattia.
Fonte: Springer