Polmonite Linfocitica che Diventa Linfoma: Un Incredibile Caso di Trasformazione Polmonare!
Amici appassionati di medicina e dei misteri che il nostro corpo a volte ci riserva, oggi voglio parlarvi di un caso davvero particolare, uno di quelli che ci ricorda quanto sia complessa e sorprendente la biologia umana. Immaginate due malattie polmonari rare, talmente rare che sentirne parlare è già un evento: la polmonite linfocitica interstiziale idiopatica (iLIP) e il linfoma MALT polmonare. Ora, immaginate che una possa, in determinate circostanze, trasformarsi nell’altra. Sembra la trama di un thriller medico, vero? Eppure, è esattamente quello che è successo in un caso che ha attirato la mia attenzione.
Due Protagoniste Rare: iLIP e Linfoma MALT
Prima di addentrarci nella storia, facciamo un piccolo ripasso. La iLIP, o polmonite linfocitica interstiziale idiopatica, è una condizione benigna, ma non per questo da sottovalutare. Si caratterizza per un’infiltrazione diffusa di linfociti (un tipo di globuli bianchi) nel tessuto polmonare. “Idiopatica” significa che non se ne conosce una causa scatenante precisa, come malattie autoimmuni o infezioni, che a volte possono provocarla. È una di quelle diagnosi che richiede un attento monitoraggio, proprio perché, come vedremo, nasconde un potenziale “lato oscuro”.
Dall’altra parte abbiamo il linfoma MALT polmonare. Questo è un tipo di linfoma non-Hodgkin, specificamente un linfoma extranodale della zona marginale (EMZL). “MALT” sta per “tessuto linfoide associato alle mucose”. Questi linfomi possono insorgere in diverse parti del corpo dove è presente questo tessuto, come lo stomaco, gli occhi, e appunto, i polmoni. Quando si sviluppa primariamente nei polmoni, parliamo di Linfoma Polmonare Primitivo (PPL), una vera rarità, rappresentando meno dell’1% di tutti i linfomi e meno dello 0.5% di tutti i tumori polmonari primitivi.
La cosa affascinante, e un po’ inquietante, è che la letteratura scientifica suggerisce che la iLIP possa, in alcuni casi, evolvere malignamente trasformandosi proprio in un linfoma MALT. Ed è qui che la nostra storia comincia.
La Storia di una Paziente: Un Viaggio Lungo 9 Anni
Immaginate una donna di 36 anni, asintomatica, che durante un controllo di routine scopre delle ombre anomale sulla sua radiografia toracica. Gli esami successivi, inclusa una TAC, confermano la presenza di queste ombre in entrambi i polmoni. Viene eseguita una broncoscopia con biopsia, che rivela un’infiltrazione linfocitaria della mucosa bronchiale. Non si può escludere un linfoma, quindi si procede con una biopsia chirurgica tramite VATS (Video-Assisted Thoracoscopic Surgery). L’esame istopatologico mostra una densa infiltrazione di piccoli linfociti, con setti interlobulari e alveolari ispessiti e alterazioni cistiche. Non emergono malattie autoimmuni o altri coinvolgimenti al di fuori del torace. La diagnosi? iLIP idiopatica.
La paziente inizia una terapia steroidea, che però interrompe, e viene quindi seguita con controlli periodici. Passano ben nove anni. Nove lunghi anni in cui la situazione sembra sotto controllo, finché una delle lesioni, quella nel lobo superiore sinistro, inizia gradualmente ad ingrandirsi. La paziente, ora 45enne, viene nuovamente indirizzata all’ospedale.
Una nuova TAC conferma l’ingrandimento della lesione e un’estesa atelettasia (collasso del tessuto polmonare) nel lobo superiore sinistro. Una nuova broncoscopia non riesce a fornire materiale sufficiente per una diagnosi certa. Data la situazione e l’inutilità funzionale del lobo colpito, si decide per un intervento chirurgico: una lobectomia superiore sinistra.
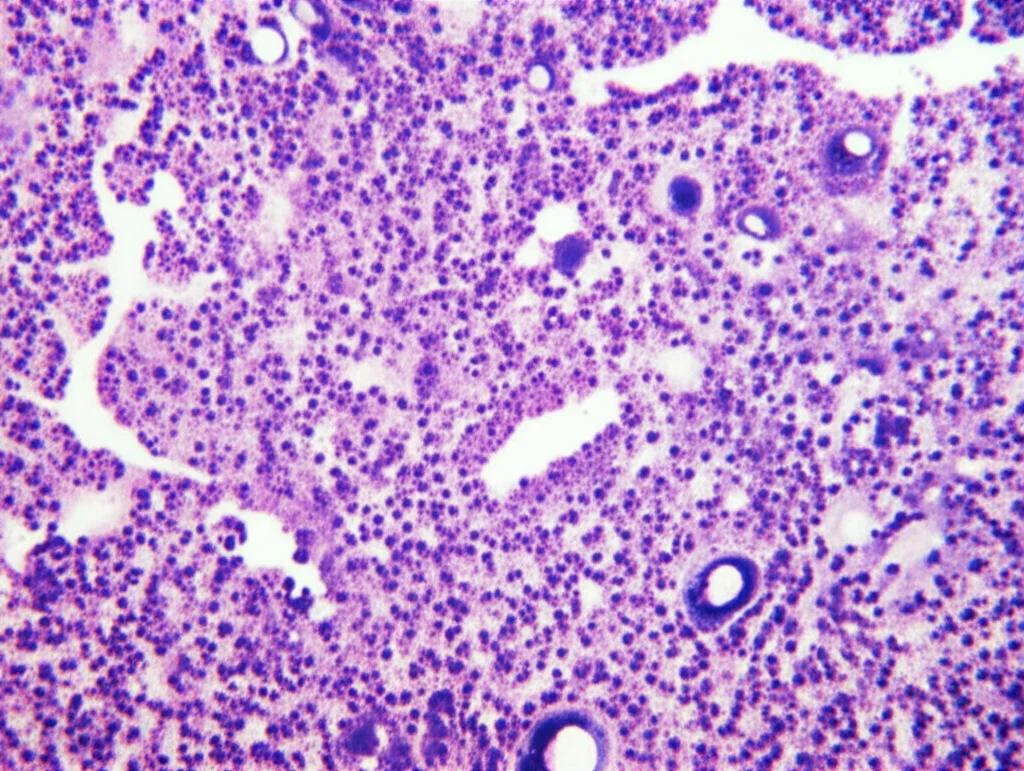
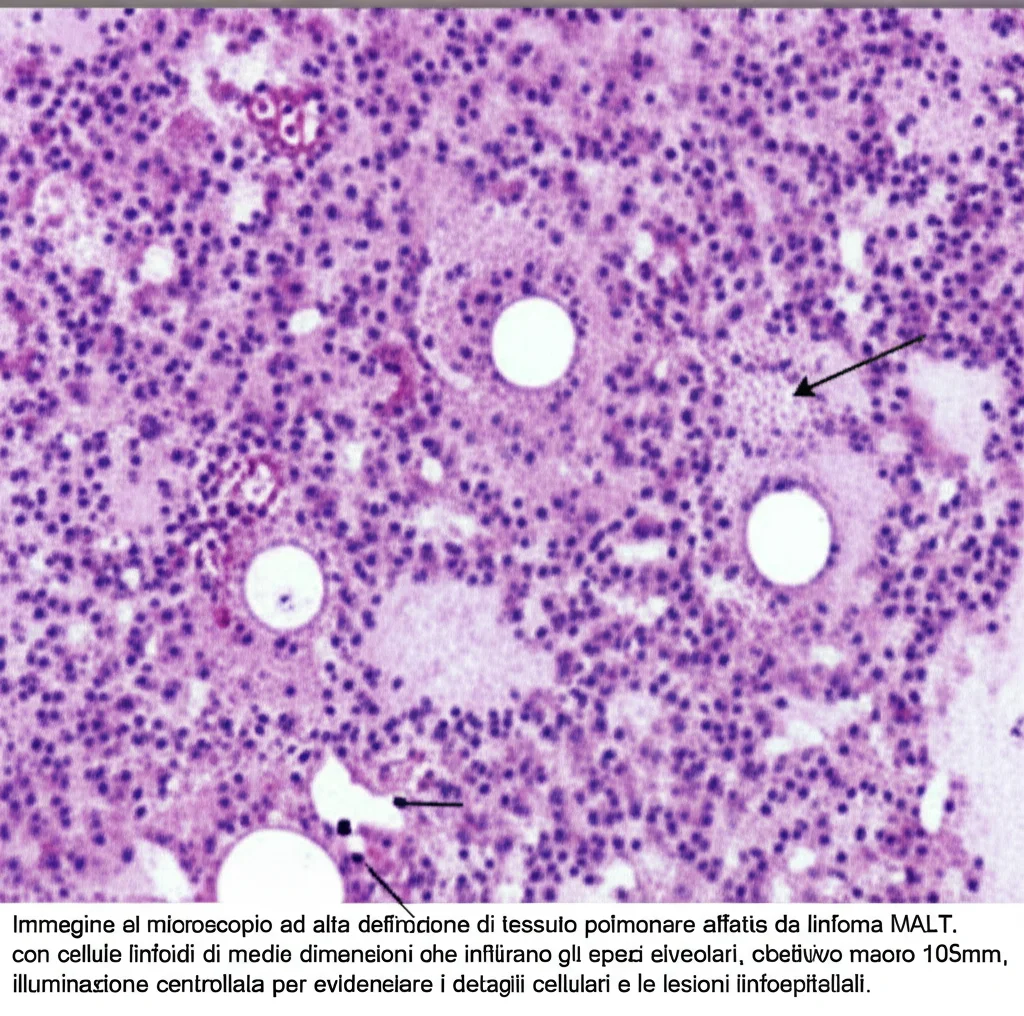
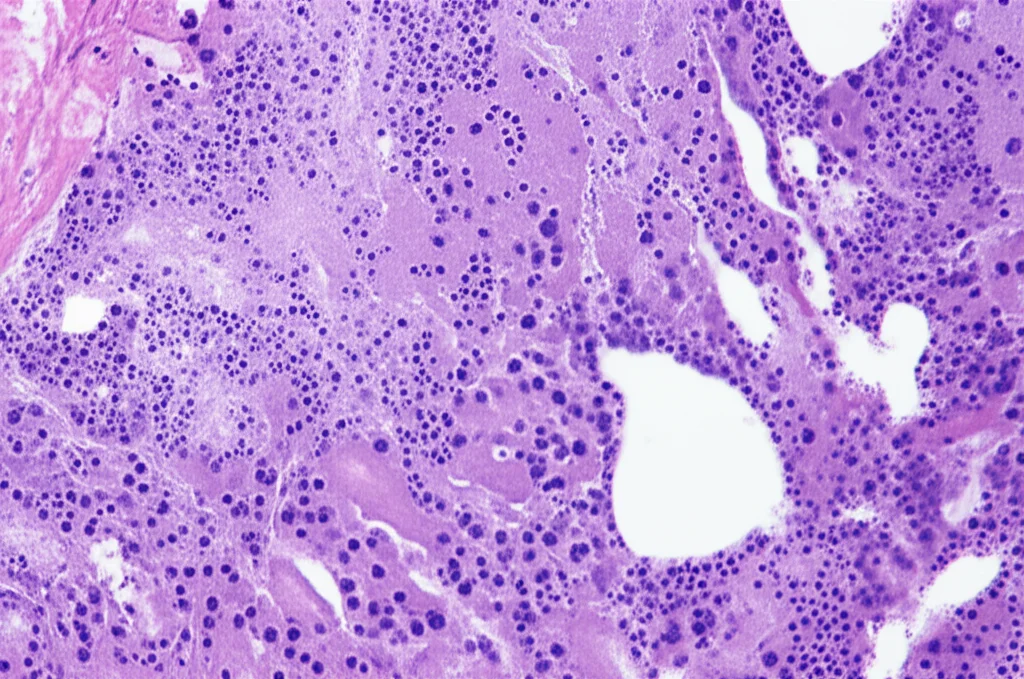
L’esame istopatologico del pezzo operatorio questa volta racconta una storia diversa. Si osserva una proliferazione diffusa di linfociti di medie dimensioni, simili a centrociti, che riempiono gli spazi alveolari. Sono presenti anche le cosiddette lesioni linfoepiteliali (LEL), un segno caratteristico. L’immunoistochimica (una tecnica che usa anticorpi per identificare specifiche molecole nelle cellule) mostra che queste cellule sono positive per CD20, CD79a e BCL-2, e negative per CD3, CD5, CD10 e ciclina D1. Confrontando questi risultati con la biopsia di nove anni prima, emerge chiaramente la differenza: i linfociti CD20 positivi ora sono di medie dimensioni, non piccoli, e le lesioni linfoepiteliali sono evidenti. La diagnosi è chiara: linfoma MALT polmonare (EMZL).
Le Sfide della Diagnosi: Un Vero Grattacapo
Questo caso sottolinea una difficoltà enorme: distinguere tra iLIP e linfoma MALT basandosi solo sui sintomi clinici (spesso assenti o aspecifici) e sui reperti radiologici è una vera sfida. Entrambe le condizioni possono presentarsi con ombre, consolidamenti o opacità a vetro smerigliato. Per questo, la biopsia è fondamentale.
Le tecniche possono variare:
- Biopsia transbronchiale: meno invasiva, ma spesso il campione è troppo piccolo o danneggiato (artefatti da schiacciamento) per una diagnosi definitiva.
- Agoaspirato TC-guidato: simile alla precedente per quanto riguarda le possibili limitazioni.
- Resezione chirurgica: più invasiva, ma fornisce campioni di tessuto più grandi e di migliore qualità, spesso necessari per una diagnosi certa.
Recentemente, la criobiopsia sta emergendo come una tecnica promettente, in grado di fornire campioni di qualità superiore senza artefatti da schiacciamento, e potrebbe diventare un approccio diagnostico preferenziale.
Dal punto di vista istopatologico, le differenze ci sono, ma bisogna saperle cercare. Nella iLIP, l’infiltrato è generalmente polimorfo, con piccoli linfociti mescolati a plasmacellule e macrofagi, e predominano i linfociti T (CD3+). Nel linfoma MALT, invece, le cellule neoplastiche sono linfociti B (CD20+, CD79a+) di medie dimensioni, che infiltrano l’epitelio bronchiale formando le LEL.
Trattamento e Prospettive: Cosa Ci Aspetta?
Una volta diagnosticato il linfoma MALT polmonare, come si procede? Il trattamento dipende dallo stadio e dalla localizzazione. Per le lesioni localizzate, la resezione chirurgica è un’opzione terapeutica, come nel caso della nostra paziente. Tuttavia, studi recenti suggeriscono che, data la natura spesso indolente (a crescita lenta) del PPL, anche un approccio di “vigile attesa” (osservazione senza trattamento attivo immediato) può essere una strategia valida, senza impattare negativamente sulla sopravvivenza o sulla prognosi. Questo può essere particolarmente vantaggioso per i pazienti asintomatici, evitando trattamenti non necessari.
Nel caso specifico, dopo l’intervento, non è stata somministrata alcuna terapia adiuvante. Purtroppo, due anni dopo, è stata rilevata una recidiva a livello gastrico. La paziente è stata quindi sottoposta a chemioterapia, ottenendo una remissione completa. A distanza di 5 anni dall’intervento chirurgico iniziale, la paziente era ancora in vita e libera da malattia da 3 anni dopo la chemioterapia.
È importante sottolineare che il rischio di recidiva per i linfomi EMZL extragastrici è riportato come elevato, e questo caso lo conferma. Non esiste un periodo di osservazione standardizzato o un trattamento aggiuntivo universalmente accettato per il linfoma MALT extragastrico, ma è cruciale un attento follow-up.
Cosa Impariamo da Questo Caso?
La trasformazione maligna da iLIP a linfoma MALT è un evento raro, ma possibile. Questo caso ci insegna diverse cose:
- L’importanza di un follow-up a lungo termine per pazienti con diagnosi di iLIP.
- Le difficoltà diagnostiche nel distinguere queste due entità, che spesso richiedono approcci bioptici più invasivi per una diagnosi definitiva.
- Anche se la chirurgia è spesso scelta sia per la diagnosi che per il trattamento, l’opzione di una diagnosi tramite biopsia meno invasiva seguita da un piano di trattamento che può includere l’osservazione è una strada percorribile.
- Nonostante la malattia possa progredire e richiedere un trattamento, una strategia iniziale basata sull’osservazione non sembra peggiorare il decorso clinico dei pazienti e può risparmiare trattamenti superflui, specialmente nei casi asintomatici.
Insomma, una storia che ci ricorda come la medicina sia un campo in continua evoluzione e come ogni paziente rappresenti un universo da esplorare con attenzione e dedizione. La consapevolezza di queste rare evenienze è cruciale per noi medici, per poter offrire ai nostri pazienti la migliore assistenza possibile.
Fonte: Springer





