
DNA, un Gene e il Rischio di Cancro al Pancreas: La Storia Affascinante di KLHL17
Ciao a tutti! Oggi voglio portarvi con me in un viaggio affascinante nel mondo della genetica e della ricerca sul cancro, in particolare quello al pancreas. È una malattia terribile, lo sappiamo, una delle principali cause di morte per cancro negli Stati Uniti e con tassi di sopravvivenza ancora troppo bassi. Ma la scienza non si ferma, e ogni giorno cerchiamo di capire meglio cosa scatena questa malattia per trovare nuove armi per combatterla.
Il DNA come Mappa del Tesoro (o del Rischio)
Immaginate il nostro DNA come un’immensa mappa. A volte, piccole variazioni in questa mappa, chiamate varianti genetiche (o polimorfismi a singolo nucleotide, SNP, se vogliamo essere tecnici), possono aumentare o diminuire il rischio di sviluppare certe malattie. Per il cancro al pancreas (specificamente l’adenocarcinoma duttale pancreatico, PDAC, che è la forma più comune), sappiamo che sia varianti rare ad alto rischio sia varianti comuni a basso rischio giocano un ruolo.
Grazie a studi enormi chiamati Studi di Associazione Genome-Wide (GWAS), in cui si confronta il DNA di migliaia di persone malate con quello di persone sane, abbiamo già identificato diverse zone del genoma associate a un rischio maggiore di PDAC. È come trovare delle “X” sulla mappa che indicano aree sospette.
Zoomando su un Indizio Promettente: chr1p36.33
Una di queste aree “sospette” si trova sul cromosoma 1, in una regione chiamata chr1p36.33. Inizialmente, uno studio l’aveva segnalata come “suggestiva”, ma unendo ancora più dati (parliamo di oltre 11.000 casi e 17.000 controlli!), il segnale è diventato forte e chiaro. La “bandierina” iniziale era uno SNP chiamato rs13303010.
Ma qui arriva il bello (e il difficile) della ricerca: un segnale GWAS è spesso solo l’inizio. Quella “X” sulla mappa non indica una singola casa, ma un intero quartiere! Ci sono tantissime varianti genetiche vicine tra loro e fortemente correlate, e capire quale sia la vera “colpevole” e come agisce richiede un lavoro da detective.
Il Lavoro da Detective: Mappatura Fine e Test Funzionali
Quindi, cosa abbiamo fatto? Ci siamo messi a “mappare finemente” questa regione. Usando metodi statistici avanzati (come SuSiE, un approccio Bayesiano) e analizzando come le varianti sono legate tra loro (linkage disequilibrium, LD), abbiamo ristretto il campo a un gruppetto di sette varianti candidate.
Il passo successivo? Capire se queste varianti fanno davvero qualcosa. La maggior parte delle varianti GWAS si trova in regioni non codificanti del DNA, quelle che non diventano proteine ma che agiscono come “interruttori” per regolare l’accensione o lo spegnimento dei geni. Abbiamo controllato se le nostre sette candidate si trovassero in zone “attive” del genoma nelle cellule pancreatiche, usando dati su come la cromatina è organizzata (ChromHMM) e quali regioni sono accessibili (ATAC-seq). E bingo! Tutte e sette si trovavano in regioni che sembravano proprio degli interruttori genetici (promotori, enhancer).
Per vedere se queste varianti influenzavano il legame di proteine regolatrici (i fattori di trascrizione), abbiamo usato una tecnica chiamata EMSA (Electrophoretic Mobility Shift Assay). In pratica, mettiamo un pezzetto di DNA contenente la variante (in entrambe le sue versioni, o alleli) a contatto con proteine estratte da cellule di cancro al pancreas. Se una proteina si lega, il DNA “rallenta” la sua corsa su un gel. Ebbene, tre delle sette varianti (rs13303010, rs13303327 e rs13303160) hanno mostrato un legame preferenziale per uno dei due alleli. Interessante!
Ma legarsi è una cosa, regolare l’espressione genica è un’altra. Abbiamo quindi usato dei test con luciferasi. In questi test, inseriamo il pezzetto di DNA con la variante davanti a un gene che produce luce (luciferasi) e vediamo quanta luce viene prodotta nelle cellule. Questo ci dice se la nostra sequenza agisce da promotore (accende il gene) o da enhancer (ne potenzia l’accensione) e se c’è differenza tra i due alleli.
Risultato? Mentre rs13303010 (la bandierina iniziale) non mostrava grandi differenze tra gli alleli, sia rs13303327 che rs13303160 hanno mostrato una chiara attività regolatoria allele-specifica. In particolare, rs13303160 si è dimostrata un potente enhancer, con l’allele protettivo (A) che “accendeva” la luciferasi molto più dell’allele di rischio (G).
Identikit dei “Complici”: i Fattori di Trascrizione JunB e JunD
Ok, abbiamo una variante “funzionale”, rs13303160, che agisce come un interruttore la cui intensità dipende dall’allele presente. Ma chi preme questo interruttore? Quali proteine si legano in modo diverso ai due alleli?
Analisi al computer (in silico) ci hanno suggerito che la famiglia di fattori di trascrizione AP-1 (che include proteine come Jun e Fos) potesse essere coinvolta. L’allele di rischio (G) sembrava disturbare il sito di legame per AP-1.
Abbiamo fatto altri esperimenti EMSA, questa volta usando proteine AP-1 purificate (ricombinanti) o stimolando le cellule con una sostanza (TPA) che attiva AP-1. E abbiamo trovato i colpevoli! Le proteine JunB e JunD si legavano in modo preferenziale all’allele protettivo (A) di rs13303160, sia in vitro (con le proteine purificate) sia nel contesto cellulare (con estratti nucleari).
Per la prova definitiva, abbiamo usato la ChIP-qPCR (Chromatin Immunoprecipitation seguita da PCR quantitativa). Questa tecnica ci permette di “pescare” le proteine legate al DNA direttamente dentro le cellule e vedere a quale allele si legano di più. E anche qui, conferma: nelle cellule di cancro al pancreas eterozigoti per rs13303160 (cioè con entrambi gli alleli A e G), JunB e JunD si legavano molto di più all’allele protettivo A rispetto all’allele di rischio G.
Quindi, abbiamo il nostro meccanismo: rs13303160 è la variante funzionale, e la sua influenza sul rischio di cancro al pancreas passa attraverso un legame differenziale dei fattori di trascrizione JunB e JunD.

Il Bersaglio Finale: il Gene KLHL17
Ma quale gene viene regolato da questo interruttore rs13303160 e dai suoi controllori JunB/D? La regione chr1p36.33 è affollata di geni. Per capirlo, abbiamo guardato ai dati eQTL (expression Quantitative Trait Locus) dal progetto GTEx, che collegano le varianti genetiche ai livelli di espressione dei geni in diversi tessuti, incluso il pancreas.
L’analisi ha puntato il dito su due geni principali: NOC2L e, soprattutto, KLHL17. L’allele di rischio (G) di rs13303010 (e quindi anche di rs13303160, visto che sono molto legati) era associato a livelli più bassi di espressione di KLHL17 nel pancreas. Analisi di colocalizzazione e studi precedenti (TWAS) rafforzavano l’idea che fosse proprio KLHL17 il gene bersaglio rilevante per il rischio di PDAC.
Che Lavoro Fa KLHL17 nel Pancreas?
KLHL17 appartiene a una famiglia di proteine (Kelch) che spesso agiscono come “adattatori”: riconoscono proteine specifiche e le portano a un complesso chiamato Cullin3-E3 ubiquitin ligase (CRL3). Questo complesso è come una squadra di smaltimento cellulare: attacca una piccola etichetta (l’ubiquitina) alle proteine bersaglio, segnalandole per la distruzione.
Per confermare che KLHL17 facesse questo anche nelle cellule pancreatiche, abbiamo fatto esperimenti di immunoprecipitazione (IP) seguiti da spettrometria di massa (MS). In pratica, abbiamo “pescato” KLHL17 dalle cellule e visto cosa si portava dietro. E sì, abbiamo trovato Cullin-3 e altri membri della squadra CRL3, confermando il suo ruolo canonico.
Ma quali proteine “etichetta” KLHL17 per la distruzione nel pancreas? L’analisi proteomica ha identificato diversi candidati. Tra i più interessanti c’erano due proteine del citoscheletro: la vimentina e la nestina. Perché interessanti? Perché entrambe sono note per essere coinvolte in processi chiave all’inizio dello sviluppo del cancro al pancreas, come l’infiammazione, la metaplasia acinare-duttale (ADM) – una trasformazione delle cellule pancreatiche che può preludere al cancro – e la transizione epitelio-mesenchimale (EMT), un processo che rende le cellule più mobili e invasive. Inoltre, i livelli di vimentina e nestina tendono ad aumentare nel PDAC. L’idea è che KLHL17 possa aiutare a tenere sotto controllo i livelli di queste due proteine.
Mettiamo Insieme i Pezzi: KLHL17, Infiammazione e Rischio di Cancro
Abbiamo provato a vedere se manipolare i livelli di KLHL17 influenzasse la crescita delle cellule tumorali pancreatiche in vitro. I risultati iniziali con siRNA (che “spegne” temporaneamente il gene) suggerivano un rallentamento della crescita, ma c’erano problemi di specificità (veniva colpito anche il vicino NOC2L) e di efficienza. Usando una tecnica più precisa (CRISPRi), abbiamo ridotto l’mRNA di KLHL17 ma, stranamente, i livelli della proteina non cambiavano molto, e non abbiamo visto effetti sulla crescita. Forse l’effetto visto con siRNA era dovuto a NOC2L? Possibile, e merita ulteriori indagini.
Ma allora, come si collega KLHL17 al rischio di cancro? Abbiamo provato un approccio diverso: un’analisi in silico. Abbiamo preso dati di espressione genica da campioni di pancreas normale (dal progetto GTEx) e li abbiamo divisi in due gruppi: quelli con alta espressione di KLHL17 e quelli con bassa espressione. Abbiamo poi confrontato quali altri geni erano espressi diversamente tra i due gruppi.
Il risultato è stato illuminante: nei campioni con bassa espressione di KLHL17 (ricordate? L’allele di rischio è associato a bassa espressione!), c’era un chiaro arricchimento di geni coinvolti nell’infiammazione e in percorsi infiammatori. Molti di questi percorsi risultavano “attivati”.
L’Ipotesi Finale: Un Meccanismo di Difesa Compormesso
Questo ci porta a formulare un’ipotesi affascinante sul ruolo di rs13303160 e KLHL17 nel cancro al pancreas.
Sappiamo che l’infiammazione cronica e lo stress cellulare nel pancreas possono portare ad ADM ed EMT, passi cruciali verso il cancro. Vimentina e nestina sono attori importanti in questi processi.
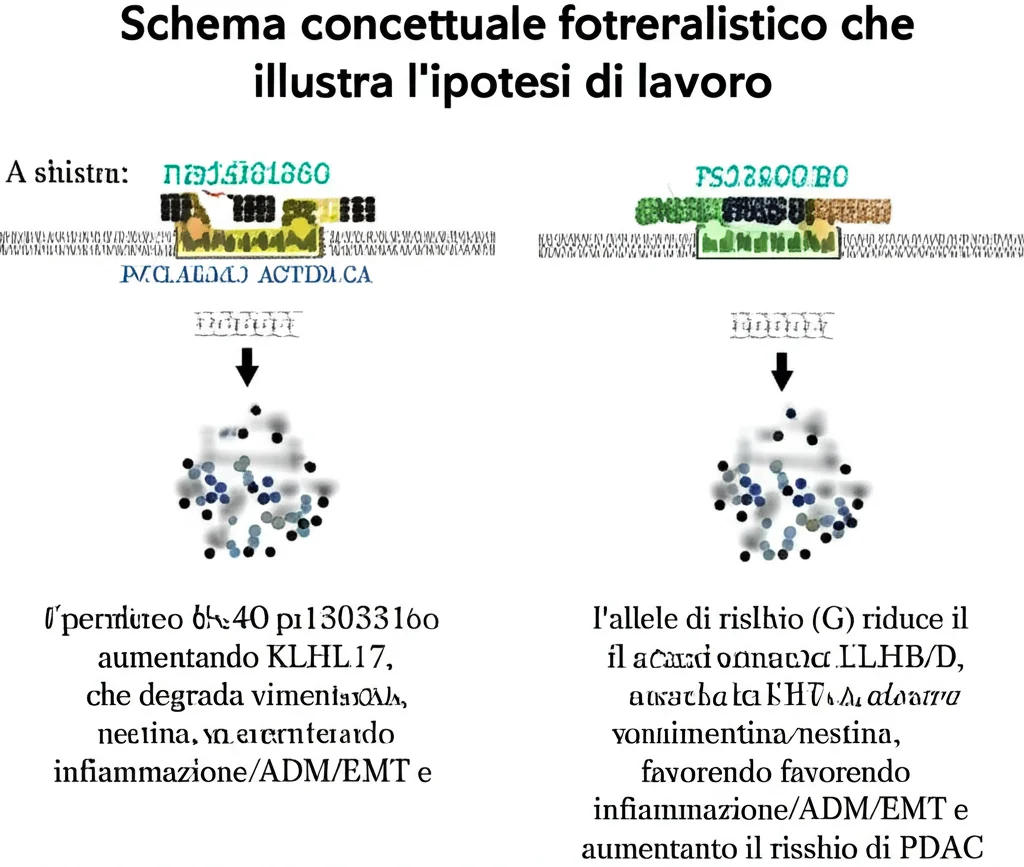
La nostra ipotesi è che KLHL17 agisca come un fattore protettivo: in condizioni di stress o infiammazione (quando JunB e JunD sono attivati), l’allele protettivo (A) di rs13303160 permette un legame efficiente di JunB/D, che a sua volta “accende” l’espressione di KLHL17. Alti livelli di KLHL17 portano alla degradazione di vimentina e nestina, aiutando a risolvere l’infiammazione e a prevenire la progressione verso ADM/EMT e quindi verso il cancro.
Al contrario, quando è presente l’allele di rischio (G), il legame di JunB/D è meno efficiente. Anche in presenza di stress, l’espressione di KLHL17 non aumenta a sufficienza. Di conseguenza, vimentina e nestina non vengono degradate efficacemente, l’infiammazione persiste, ADM ed EMT sono favorite, e il rischio di sviluppare il cancro al pancreas aumenta.
È una storia complessa, vero? Ma è incredibile come lo studio di una singola lettera nel nostro DNA possa aprirci finestre su meccanismi biologici così intricati e rilevanti per una malattia devastante. Certo, questa è un’ipotesi basata su molti esperimenti in vitro e analisi in silico. Serviranno ulteriori studi, soprattutto in vivo, per confermare questo meccanismo e capire appieno il ruolo di KLHL17 nel pancreas e nello sviluppo del cancro. Ma ogni passo avanti nella comprensione è fondamentale per sviluppare future strategie di prevenzione e terapia. Spero che questo viaggio nella genetica del cancro al pancreas vi abbia affascinato quanto ha affascinato me!
Fonte: Springer





