Istiocitosi a Cellule di Langerhans: Viaggio nel Cuore di una Malattia Rara Pediatrica
Ciao a tutti! Oggi voglio portarvi con me in un viaggio affascinante, anche se a tratti complesso, nel mondo di una malattia rara chiamata Istiocitosi a Cellule di Langerhans (LCH), concentrandoci in particolare su come si manifesta nei bambini. È una condizione che può sembrare un vero rompicapo per medici e famiglie, proprio per la sua rarità e per come può presentarsi in modi molto diversi.
Prendiamo spunto da un caso specifico, quello di un ragazzo di 13 anni, per capire meglio di cosa parliamo. Ma prima, facciamo un passo indietro.
Cos’è l’Istiocitosi a Cellule di Langerhans?
Immaginate delle cellule speciali, le cellule di Langerhans, che normalmente fanno parte del nostro sistema immunitario. Nell’LCH, queste cellule iniziano a moltiplicarsi in modo anomalo. Non sono cellule “cattive” nel senso tumorale classico, ma la loro crescita eccessiva e il loro accumulo in diversi organi (come ossa, pelle, linfonodi, fegato, polmoni…) creano infiammazione e danni.
È considerata una malattia rara, soprattutto nei bambini. Pensate che colpisce circa 5-9 bambini su un milione sotto i 15 anni, con un’età media alla diagnosi intorno ai 3 anni. Negli adulti è ancora più rara.
La cosa che la rende così sfidante è la sua eterogeneità:
- Può presentarsi con una singola lesione (unifocale).
- Può coinvolgere più punti dello stesso sistema (es. più ossa – multifocale a singolo sistema).
- Può colpire più sistemi d’organo contemporaneamente (multisistemica), che è la forma potenzialmente più seria.
I sintomi sono altrettanto vari: dolore osseo, eruzioni cutanee, febbre, perdita di appetito e peso, stanchezza. La prognosi dipende molto dall’estensione della malattia e dal coinvolgimento di organi considerati “a rischio” (come fegato, milza, midollo osseo).
Un Caso Esemplare: La Storia di un Giovane Paziente
Torniamo al nostro ragazzo di 13 anni. Si presenta al pronto soccorso con dolore e gonfiore all’orecchio destro da alcuni giorni. Niente febbre alta, nessuna storia medica particolare, vaccinazioni in regola. I familiari gli avevano dato un antibiotico (amoxicillina/clavulanato) per 10 giorni, ma senza risultati. Un dettaglio curioso: usava cuffie antirumore per dormire.
All’esame fisico, si nota questo gonfiore dolente nella regione temporale (circa 4 cm) e l’otoscopia mostra un timpano scuro. Gli esami del sangue sono più o meno nella norma, a parte una lieve alterazione della proteina C reattiva (PCR), indice di infiammazione.
Qui entra in gioco l’imaging. Un’ecografia mostra una formazione ipoecogena (cioè che riflette poco gli ultrasuoni, spesso liquida o semi-solida) di circa 4.5 cm sopra l’orecchio destro, compatibile con una raccolta. Ma non solo: l’osso temporale sottostante appare irregolare, quasi “mangiucchiato”, con piccoli frammenti. Si nota anche un linfonodo sospetto nella ghiandola parotide vicina e altri linfonodi ingrossati all’angolo della mandibola.
Si procede con una TAC (Tomografia Computerizzata). Il cervello è a posto, ma la lesione all’osso temporale e al mastoide (la parte ossea dietro l’orecchio) è ben visibile, con un’estensione anche verso l’interno del cranio (probabilmente epidurale, cioè sopra la dura madre) e verso i tessuti molli esterni. La densità è fluida/sovrafluida, con un bordo leggermente più denso. L’osso è chiaramente interrotto e rarefatto. Anche la cassa timpanica e le cellule mastoidee sono piene di liquido.
A questo punto, il sospetto di qualcosa di più complesso di una semplice infezione si fa forte. Il ragazzo viene trasferito in un centro specializzato dove una Risonanza Magnetica (MRI) conferma i reperti. Si decide per un intervento chirurgico per rimuovere la lesione. Sorpresa: non si trova pus, ma tessuto infiammatorio. Questo fa scattare il campanello d’allarme per l’istiocitosi.
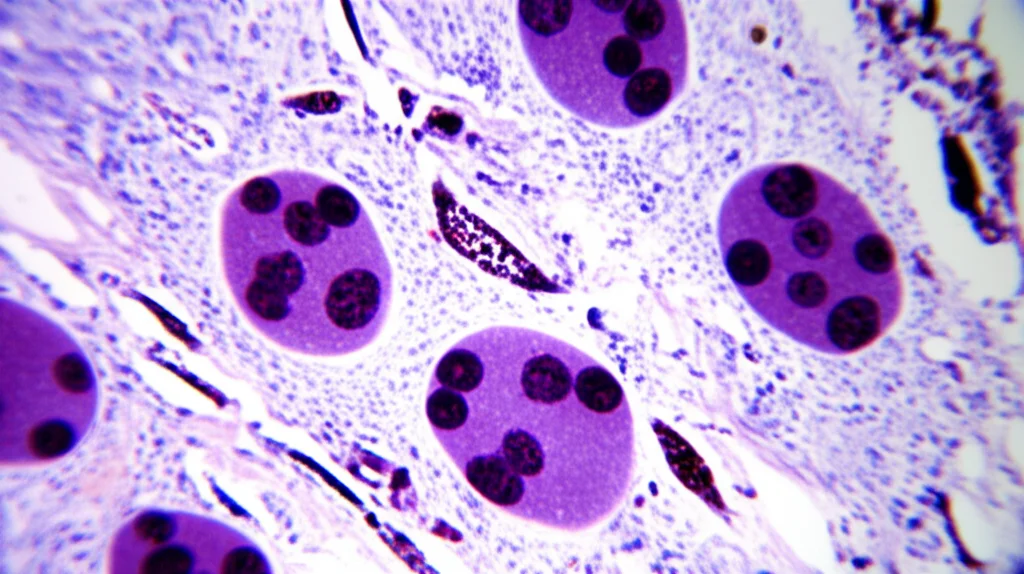
La biopsia del tessuto rimosso è la chiave. Al microscopio si vedono numerose cellule simili a istiociti (un tipo di cellula immunitaria) che proliferano. Hanno caratteristiche specifiche: citoplasma abbondante e rosato (eosinofilo), nuclei dalla forma irregolare, quasi “a chicco di caffè”, con delle scanalature. Intorno a queste cellule, un’infiltrazione di altre cellule infiammatorie, soprattutto eosinofili.
L’ultimo passo è l’immunoistochimica: si usano anticorpi specifici per “colorare” le cellule e identificarle. Le cellule sospette risultano positive per CD1a e proteina S100, marcatori tipici delle cellule di Langerhans. La diagnosi è confermata: Istiocitosi a Cellule di Langerhans multisistemica, con coinvolgimento dell’osso temporale, della parotide e dei linfonodi.
La Sfida della Diagnosi: Un Vero Rompicapo Medico
Questo caso ci mostra quanto possa essere difficile arrivare alla diagnosi. L’LCH è rara, i sintomi possono essere vaghi o mimare altre condizioni (infezioni, altre malattie infiammatorie, persino tumori). L’imaging è fondamentale per vedere l’estensione del problema, ma spesso non è specifico. L’ecografia, la TAC, la RMN danno indizi importanti, ma a volte è necessaria anche la PET (Tomografia a Emissione di Positroni) con [18F]-FDG, che mostra l’attività metabolica dei tessuti e aiuta a stadiare la malattia e monitorare la risposta alla terapia.
La conferma definitiva, però, arriva quasi sempre dalla biopsia con l’analisi istologica e immunoistochimica. È l’identificazione di quelle cellule specifiche con i marcatori CD1a, S100 e CD207 (langerina) che toglie ogni dubbio.
Dietro le Quinte: La Genetica dell’LCH
Negli ultimi anni abbiamo capito molto di più sulla biologia dell’LCH. Si è scoperto che, nella maggior parte dei casi, le cellule dell’LCH presentano mutazioni genetiche specifiche, in particolare nella via di segnalazione cellulare chiamata MAPK (Mitogen-Activated Protein Kinase). La mutazione più comune (circa 50-65% dei casi) è la BRAF V600E, spesso associata a forme più severe e multisistemiche.
Questa scoperta ha cambiato la prospettiva: l’LCH non è più vista solo come un disordine infiammatorio, ma come una vera e propria neoplasia mieloide, cioè un tumore derivante da precursori delle cellule del sangue nel midollo osseo. Questo apre la strada a terapie mirate. Recentemente sono state identificate anche varianti genetiche germinali (ereditarie), come quelle nel gene SMAD6, che potrebbero aumentare la suscettibilità all’LCH.
Affrontare il Nemico: Le Strategie Terapeutiche
Come si cura l’LCH? Dipende dalla forma. Le forme localizzate, magari solo cutanee, a volte regrediscono spontaneamente o richiedono trattamenti solo se sintomatiche. Le forme multifocali o multisistemiche, come quella del nostro paziente, richiedono un trattamento sistemico.
Lo standard attuale, seguito anche nel caso descritto (protocollo LCH4), prevede una combinazione di chemioterapia (tipicamente Vinblastina) e corticosteroidi (come il Prednisolone) per circa 12 mesi. L’intensità della terapia viene adattata alla gravità:
- Casi “lievi” (con sintomi prevalentemente infiammatori) rispondono bene a regimi a bassa intensità.
- Casi “severi” (con sintomi simil-tumorali o coinvolgimento di organi a rischio) richiedono terapie più intensive, che sono efficaci ma possono avere più effetti collaterali (come perdita di capelli, tossicità ematologica).
Per i casi refrattari (che non rispondono) o recidivanti (che ritornano dopo il trattamento), esistono terapie di “salvataggio”, spesso con farmaci come Citarabina, Vindesina e Prednisone. Un fattore prognostico chiave è la risposta precoce al trattamento iniziale.
Nel nostro caso, il ragazzo ha risposto significativamente bene alla terapia con Prednisolone e Vinblastina per 12 mesi, il che è una notizia fantastica e dimostra l’efficacia dei protocolli attuali.
Vaccinazioni e LCH: C’è un Legame?
Una domanda che a volte emerge è se ci sia un legame tra le vaccinazioni e l’insorgenza dell’LCH. Attualmente, non ci sono prove solide che stabiliscano una connessione causale. È stato riportato un caso interessante di iperplasia (aumento benigno) di cellule di Langerhans in un linfonodo dopo una vaccinazione COVID-19, che inizialmente aveva fatto sospettare l’LCH. Tuttavia, si è poi rivelata una reazione immunitaria benigna e transitoria, non la malattia vera e propria.
Questo ci ricorda che il sistema immunitario può reagire in modi complessi e che è importante distinguere le reazioni fisiologiche dalle malattie neoplastiche. Servono ulteriori ricerche per capire appieno il ruolo del sistema immunitario nello sviluppo dell’LCH, ma al momento le vaccinazioni di routine non sono considerate un fattore di rischio.
Uno Sguardo al Futuro: Ricerca e Nuove Speranze
La ricerca sull’LCH è in continua evoluzione. Le priorità future sono chiare:
- Approfondire la genetica: Continuare a identificare mutazioni (come BRAF V600E e altre nella via MAPK) e capire come correlano con l’andamento clinico. Studiare le varianti germinali (come SMAD6) per comprendere la suscettibilità ereditaria.
- Sviluppare terapie mirate: L’identificazione delle mutazioni apre la porta a farmaci “intelligenti” che colpiscono specificamente quelle alterazioni. Gli inibitori di MEK (un componente della via MAPK), come il cobimetinib, si sono già dimostrati promettenti nei casi refrattari. Si stanno esplorando anche altri bersagli, come la proteina SIRPα.
- Personalizzare il trattamento: Integrare i dati genetici con le caratteristiche cliniche per scegliere la terapia più adatta a ogni singolo paziente, migliorando l’efficacia e riducendo la tossicità.
L’LCH rimane una sfida, una malattia rara che richiede un approccio multidisciplinare (pediatri, oncologi, radiologi, patologi, chirurghi…). Ma la conoscenza sta crescendo rapidamente. Ogni caso, come quello del ragazzo di 13 anni, ci insegna qualcosa e ci spinge a cercare risposte migliori. La combinazione di ricerca genetica, terapie innovative e un’attenta gestione clinica offre una speranza concreta per migliorare la vita dei giovani pazienti affetti da questa condizione complessa.
Fonte: Springer