
SLA: Bloccare i Trimeri di SOD1, la Nuova Frontiera della Ricerca?
Ciao a tutti, appassionati di scienza e curiosi! Oggi voglio parlarvi di un argomento che mi sta particolarmente a cuore e che potrebbe rappresentare una svolta pazzesca nella lotta contro una malattia terribile: la Sclerosi Laterale Amiotrofica, meglio conosciuta come SLA. Preparatevi, perché stiamo per addentrarci nei meandri della ricerca più avanzata, ma cercherò di raccontarvela come se fossimo al bar a fare due chiacchiere, promesso!
La SLA: un nemico subdolo e ancora senza una vera cura
Partiamo dalle basi, per chi magari non la conoscesse a fondo. La SLA è una malattia neurodegenerativa progressiva che, detto in parole povere, colpisce i motoneuroni, quelle cellule nervose che dal cervello e dal midollo spinale controllano i nostri muscoli. Immaginatevi un interruttore che smette piano piano di funzionare: i muscoli si indeboliscono, arriva la paralisi, la rigidità, si perde la capacità di muoversi e, purtroppo, la situazione peggiora fino all’insufficienza respiratoria, che spesso è la causa del decesso. Il tutto, in genere, in un arco di tempo drammaticamente breve, 2-5 anni dalla diagnosi. Un vero incubo, ve lo assicuro.
La cosa ancora più frustrante è che, ad oggi, non abbiamo una cura definitiva. Certo, ci sono stati dei progressi, la FDA ha approvato alcuni farmaci come Riluzolo, Edaravone, e Tofersen (mentre Relyvrio è stato ritirato volontariamente dopo risultati non confermati in fase 3), ma questi agiscono principalmente sulla gestione dei sintomi, non bloccano né invertono la progressione della malattia. Capite bene che c’è un bisogno disperato, un “unmet medical need” come diciamo noi addetti ai lavori, di trattamenti nuovi ed efficaci.
Circa il 90% dei casi di SLA sono sporadici, cioè senza una causa genetica chiara, mentre il 10% sono forme familiari, ereditarie. Pensate che sono state identificate circa 150 diverse mutazioni genetiche associate alla SLA! Tra queste, una delle più studiate è quella a carico del gene SOD1.
L’aggregazione proteica: un “ingorgo” tossico per le cellule
Avete presente quando in città si crea un ingorgo pazzesco e tutto si blocca? Ecco, qualcosa di simile succede nelle malattie neurodegenerative. Proteine che normalmente svolgono funzioni utili, per qualche motivo iniziano a “ripiegarsi” male (misfolding) e ad aggregarsi, formando degli ammassi tossici per le cellule. Questo succede nell’Alzheimer con le placche di beta-amiloide e i grovigli di proteina tau, o nel Parkinson con i corpi di Lewy. Ebbene, anche nella SLA l’aggregazione proteica sembra giocare un ruolo da protagonista, e la proteina SOD1 è una delle principali indiziate.
L’aggregazione di SOD1 è stata osservata nei pazienti SLA, ma il meccanismo preciso è ancora un po’ un mistero. Addirittura, sembra che l’aggregazione di SOD1 possa essere un fattore comune sia nelle forme familiari (fALS) che in quelle sporadiche (sALS), magari innescata da altre proteine “dispettose” come la TDP-43 mal localizzata, o da condizioni di stress cellulare. Insomma, un bel pasticcio.
Il trucco sta nei trimeri? La scoperta che cambia le carte in tavola
E qui, amici, arriva il bello! Recenti studi hanno iniziato a suggerire che non sono tanto i grossi ammassi di SOD1 a essere critici nelle fasi iniziali della malattia, quanto piuttosto delle forme intermedie più piccole: i trimeri di SOD1. Un trimero, per capirci, è un complesso formato da tre molecole di SOD1 unite insieme. Sembra che questi trimeri siano particolarmente “cattivi” e capaci di indurre la morte dei neuroni. Quindi, l’idea geniale è stata: e se invece di combattere i grossi aggregati, provassimo a impedire la formazione di questi trimeri tossici?
In passato, nel nostro laboratorio, avevamo identificato un composto, chiamato PRG-A-01, che sembrava inibire l’aggregazione di SOD1, riducendo la sua tossicità e allungando la vita in modelli animali di SLA. Un buon inizio, ma PRG-A-01 aveva dei difetti: scarsa biodisponibilità (cioè, poco farmaco arrivava dove doveva agire) e veniva degradato troppo in fretta. Bisognava fare di meglio.

Così, ci siamo messi al lavoro per creare dei “derivati”, delle versioni modificate di PRG-A-01, e alla fine abbiamo tirato fuori dal cilindro un candidato che sembra davvero promettente: PRG-A-04.
PRG-A-04: un cecchino contro i trimeri di SOD1
Questo nuovo composto, PRG-A-04, ha dimostrato subito delle caratteristiche farmacocinetiche molto più interessanti, inclusa una buona biodisponibilità e, cosa importantissima, la capacità di attraversare la barriera emato-encefalica. Sapete, quella specie di “dogana” super selettiva che protegge il nostro cervello. Se un farmaco non la passa, è difficile che possa agire sulle malattie neurologiche.
Ma cosa fa esattamente PRG-A-04? Gli esperimenti in vitro (cioè in laboratorio, su cellule) ci hanno mostrato che PRG-A-04 si lega selettivamente alla forma mutata di SOD1 (quella problematica) e non a quella “sana” (wild type). E, cosa cruciale, inibisce efficacemente l’aggregazione causata da una particolare mutante di SOD1 (SOD1-G147P) che stabilizza proprio i trimeri. Questo ci ha fatto drizzare le antenne: forse PRG-A-04 agisce proprio impedendo la formazione o la stabilizzazione di questi trimeri!
Abbiamo fatto degli esperimenti fighissimi usando SOD1 marcate con colori fluorescenti diversi. Immaginatevi delle lucine blu, rosse e verdi. Quando nelle cellule veniva espressa la SOD1 mutata (ad esempio, verde), questa induceva anche le SOD1 sane (blu e rosse) ad aggregarsi tutte insieme in un unico punto. Trattando le cellule con PRG-A-04, questa co-aggregazione spariva! Questo suggerisce che la SOD1 mutata “trascina” quella sana in un processo che forma una struttura trimerica, e PRG-A-04 riesce a sventare questo piano.
Analisi più approfondite (LC-MS/MS e GST pull-down, per i più tecnici) hanno confermato che PRG-A-04 si lega direttamente alla SOD1 mutata in un rapporto di circa 1:3. Una molecola di PRG-A-04 per ogni tre di SOD1. Coincidenza? Io non credo! Sembra proprio che PRG-A-04 “intercetti” il trimero di SOD1.
Non solo aggregazione, ma anche propagazione!
Un’altra caratteristica insidiosa della SLA è che la “patologia” legata a SOD1 sembra diffondersi da una cellula all’altra, un po’ come un’infezione. Abbiamo ipotizzato che la SOD1 “malata” potesse essere “catturata” più facilmente dalle cellule neuronali rispetto a quella sana. E infatti, esperimenti con proteine SOD1 ricombinanti hanno mostrato che le cellule neuronali assorbono (tramite un processo chiamato endocitosi) più volentieri la SOD1 mutata. E indovinate un po’? Anche l’efficienza di questa “cattura” sembra dipendere dalla formazione di trimeri! Le mutanti che stabilizzano i trimeri vengono assorbite meglio, quelle che li destabilizzano, peggio.
La notizia bomba è che PRG-A-04 non solo riduce l’aggregazione all’interno della cellula, ma inibisce anche la “cattura” della SOD1 mutata dall’ambiente extracellulare, bloccando quindi anche la sua propagazione. Un doppio colpo micidiale!
PRG-A-04 alla prova dei fatti: i test sul modello animale
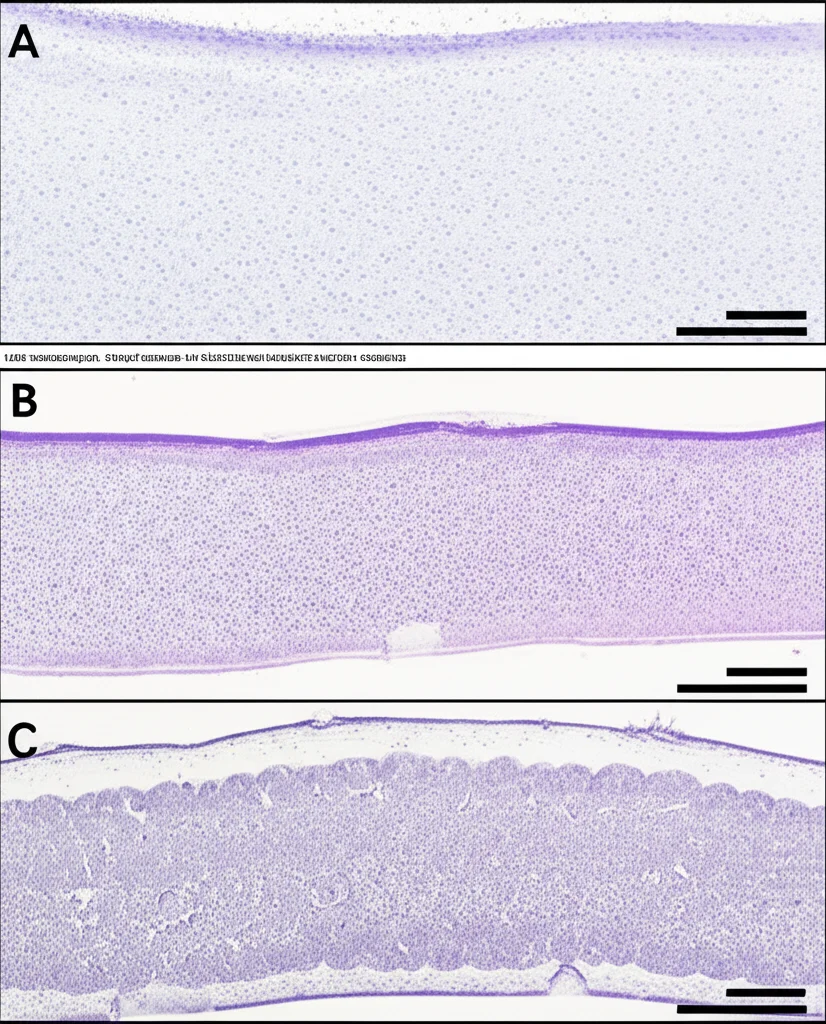
Ok, tutto molto bello in laboratorio, ma funziona anche in un organismo complesso? Per scoprirlo, abbiamo usato dei topolini modello per la SLA, i SOD1G93A-Tg, che sviluppano una malattia molto simile a quella umana. Abbiamo somministrato PRG-A-04 a questi topolini, sia per iniezione intraperitoneale che, successivamente, per via orale (una formulazione chiamata ASD, Amorphous Solid Dispersions, con una biodisponibilità di circa il 57%!).
I risultati sono stati davvero incoraggianti. La somministrazione orale di PRG-A-04 nei topolini SLA ha:
- Inibito l’aggregazione di SOD1 nel midollo spinale.
- Protetto dalla perdita di neuroni. Abbiamo visto più neuroni sani e meno segni di degenerazione.
- Migliorato la forza muscolare e la coordinazione motoria, misurate con test specifici come il rotarod (una specie di tronco rotante su cui i topi devono restare in equilibrio).
- Esteso la sopravvivenza dei topolini di circa 3 settimane! Che, rapportato alla vita di un topo, non è affatto poco.
Analisi più sofisticate, come la microarray sul tessuto del midollo spinale, hanno rivelato che PRG-A-04 riusciva a “salvare” l’espressione di geni importanti per lo sviluppo degli assoni neuronali e la stabilità delle sinapsi (i punti di contatto tra neuroni), che invece erano “spenti” nei topi malati non trattati. Inoltre, riduceva l’espressione di geni legati alla neuroinfiammazione, un altro processo dannoso nella SLA.
E la proteina TDP-43? PRG-A-04 potrebbe aiutare anche lì!
Un’altra proteina che fa spesso parlare di sé nella SLA è la TDP-43. Nel 97% dei pazienti SLA si osservano alterazioni di TDP-43, che tende a spostarsi dal nucleo (dove svolge le sue normali funzioni) al citoplasma, dove forma aggregati. Questa “fuga” di TDP-43 dal nucleo può causare problemi nella produzione di altre proteine cruciali per i neuroni, come STMN2 e UNC13A. Inoltre, gli aggregati citoplasmatici di TDP-43 sono tossici.
La cosa interessante è che, in molti pazienti con SLA sporadica, le proteine SOD1 si trovano co-localizzate con gli aggregati di TDP-43. Ebbene, i nostri esperimenti hanno mostrato che PRG-A-04 non solo riduce l’aggregazione di SOD1 indotta da un eccesso di TDP-43, ma riduce anche l’auto-aggregazione di TDP-43 e la sua localizzazione anomala nel citoplasma! Questo è importantissimo, perché suggerisce che PRG-A-04 potrebbe avere un raggio d’azione più ampio, colpendo due piccioni (SOD1 e TDP-43) con una fava, e quindi essere utile sia nelle forme familiari legate a SOD1 che in quelle sporadiche dove TDP-43 è la protagonista.
Prospettive future: cosa ci aspetta?
Ragazzi, i dati su PRG-A-04 sono davvero entusiasmanti. Abbiamo un composto che:
- Ha buone proprietà farmacologiche (biodisponibilità orale, penetrazione della barriera emato-encefalica).
- Sembra agire su un meccanismo nuovo e cruciale: l’inibizione della formazione dei trimeri di SOD1.
- Mostra efficacia nel ridurre l’aggregazione di SOD1, proteggere i neuroni, migliorare la funzione motoria e allungare la vita in modelli animali di SLA.
- Potrebbe avere effetti benefici anche sulla patologia legata a TDP-43.
Questo posiziona PRG-A-04 come un candidato farmaco molto forte per la SLA, potenzialmente utile sia per le forme familiari che per quelle sporadiche. Certo, la strada per arrivare a un farmaco per l’uomo è ancora lunga e piena di sfide. Serviranno ulteriori studi preclinici per confermare la sicurezza e poi, si spera, studi clinici sull’uomo.
Stiamo anche conducendo studi di “modeling” per capire esattamente come si formano questi trimeri di SOD1, come interagiscono con TDP-43 e in quale punto preciso del trimero si lega PRG-A-04. Più capiamo i dettagli, più potremo affinare le nostre armi contro questa malattia.
Rispetto a trattamenti esistenti, come Tofersen (che è un oligonucleotide antisenso specifico per le mutazioni di SOD1 e richiede iniezione intratecale), PRG-A-04 ha il vantaggio di poter essere somministrato per via orale e di agire su un meccanismo (la trimerizzazione) che potrebbe essere comune a diverse forme di aggregazione di SOD1, anche quella non direttamente causata da mutazioni genetiche ma da stress cellulare o dalla “cattiva compagnia” di TDP-43.
Insomma, la ricerca non si ferma mai e scoperte come questa ci danno una speranza concreta. L’idea di poter colpire la SLA alla radice, impedendo la formazione di queste strutture trimeriche tossiche, è affascinante e apre scenari terapeutici completamente nuovi. Io, da parte mia, continuerò a seguire con il fiato sospeso gli sviluppi di PRG-A-04 e di altre strategie innovative. E voi, continuate a seguirmi per restare aggiornati sulle meraviglie (e le fatiche!) del mondo della ricerca scientifica!
Alla prossima!
Fonte: Springer





