Alzheimer: Svelato un Nuovo Bersaglio! Come Bloccare ELK1 Ringiovanisce la Memoria
Ciao a tutti! Oggi voglio parlarvi di qualcosa di veramente affascinante che sta emergendo dalla ricerca sull’Alzheimer, una malattia che, purtroppo, conosciamo fin troppo bene per il suo impatto devastante sulla memoria e sulla vita delle persone anziane e delle loro famiglie. Sapete, la battaglia contro l’Alzheimer è complessa, ma ogni nuova scoperta ci avvicina a possibili soluzioni. E recentemente, abbiamo puntato i riflettori su un attore molecolare forse inaspettato: una proteina chiamata ELK1.
Alzheimer: Un Intrico di Placche e Proteine
Prima di tuffarci nel vivo della scoperta, rinfreschiamoci un attimo la memoria (ironico, vero?) su cosa succede nel cervello colpito da Alzheimer. I segni distintivi sono principalmente due: le famigerate placche senili, accumuli extracellulari di un peptide chiamato amiloide-beta (Aβ), e gli ammassi neurofibrillari intracellulari, formati dalla proteina tau iperfosforilata. Noi ci concentreremo sul primo problema: l’Aβ.
Questo peptide Aβ non spunta dal nulla. Deriva da una proteina più grande, la proteina precursore dell’amiloide (APP), che viene “tagliata” da due enzimi, come delle forbici molecolari: la β-secretasi (BACE1) e la γ-secretasi. È proprio la γ-secretasi, un complesso proteico di cui fa parte la cruciale Presenilina 1 (PS1), a completare il lavoro, liberando l’Aβ che poi si aggrega formando le placche. Tenete a mente PS1, perché è una protagonista della nostra storia.
ELK1: Un Sospettato nel Mirino
Ora, entra in scena ELK1. È un fattore di trascrizione, cioè una proteina che regola l’espressione dei geni. Normalmente viene attivato da un’altra via di segnalazione, quella di ERK1/2, che guarda caso è già nota per essere iperattiva e contribuire ai danni nell’Alzheimer. Ci siamo chiesti: che ruolo gioca ELK1 in tutto questo?
La prima cosa che abbiamo notato è stata sorprendente: i livelli della proteina ELK1 erano significativamente aumentati nei tessuti cerebrali di pazienti con Alzheimer e anche nei modelli animali della malattia (topolini geneticamente modificati per sviluppare sintomi simili all’AD). Non solo ELK1 totale, ma anche la sua forma attivata, fosforilata (p-ELK1), era più abbondante. Questo ci ha fatto drizzare le antenne: e se ELK1 fosse coinvolto direttamente nella produzione di Aβ?
La Scoperta: ELK1 Protegge PS1 dalla Degradazione
Essendo un fattore di trascrizione, la prima ipotesi era che ELK1 potesse aumentare la produzione di APP, BACE1 o PS1 agendo sui loro geni. Ma i nostri esperimenti in cellule modello hanno mostrato che non era così: ELK1 non influenzava l’RNA messaggero (l’istruzione per produrre la proteina) di questi geni.
Allora, come agisce? Abbiamo scoperto qualcosa di molto più sottile. Quando abbiamo ridotto i livelli di ELK1 nelle cellule (usando una tecnica chiamata “knockdown genetico” con shRNA), abbiamo visto che i livelli della proteina PS1 diminuivano drasticamente! Al contrario, aumentare ELK1 non cambiava molto i livelli di PS1 (forse perché nelle cellule malate ce n’è già tanto?). Questo suggeriva che ELK1 non aumentasse la produzione di PS1, ma ne impedisse la degradazione.
Come fa il nostro corpo a sbarazzarsi delle proteine vecchie o danneggiate? Principalmente attraverso due sistemi: l’autofagia-lisosomi e il sistema ubiquitina-proteasoma. Abbiamo verificato quale fosse responsabile per PS1 e abbiamo scoperto che è soprattutto il secondo: PS1 viene “etichettata” con delle molecole di ubiquitina (un processo chiamato ubiquitinazione) e poi distrutta dal proteasoma, una sorta di trita-documenti cellulare.
E qui sta il punto: abbiamo visto che ELK1 inibisce proprio l’ubiquitinazione di PS1. Meno etichette, meno distruzione, più PS1 in giro a produrre Aβ.
SYVN1: L’Esecutore Bloccato da ELK1
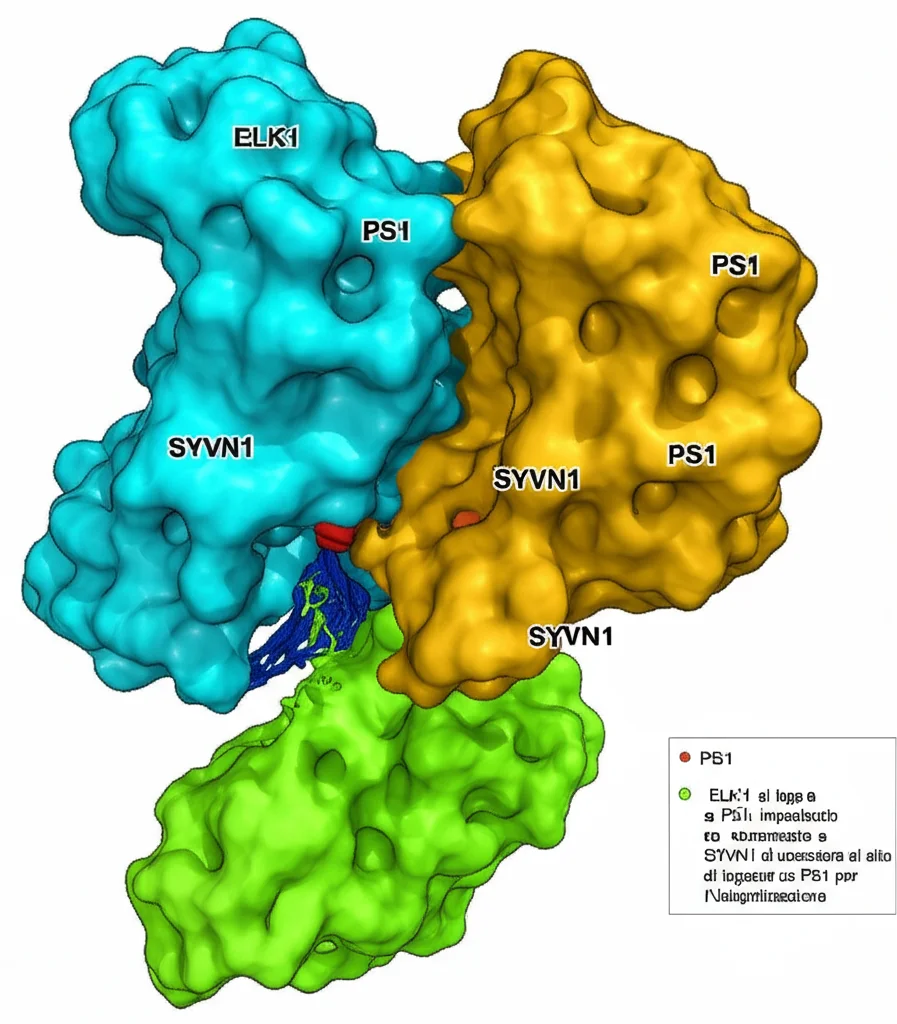
Ma chi è che normalmente appiccica queste etichette di ubiquitina a PS1? Ogni proteina ha la sua “E3 ligasi” specifica. Usando database predittivi e conferme sperimentali, abbiamo identificato il colpevole (o meglio, l’esecutore mancato): una proteina chiamata SYVN1.
Abbiamo dimostrato che:
- SYVN1 interagisce direttamente con PS1.
- Se aumentiamo SYVN1, la degradazione di PS1 accelera e la sua ubiquitinazione aumenta.
- Se blocchiamo SYVN1 con un inibitore (LS-102), i livelli di PS1 aumentano.
Quindi, SYVN1 è l’enzima che normalmente si occupa di far degradare PS1. Ma come entra in gioco ELK1? Abbiamo scoperto che ELK1 interagisce anch’esso con PS1 e, così facendo, impedisce a SYVN1 di legarsi a PS1. È come se ELK1 si mettesse in mezzo, proteggendo PS1 dall’essere etichettata per la distruzione da parte di SYVN1. E la forma fosforilata, p-ELK1, sembra essere ancora più “brava” a fare questo lavoro di interferenza!
La Prova del Nove: Bloccare ELK1 Funziona nei Topi!
Tutto molto interessante in provetta, ma funziona nel contesto di un organismo complesso? Per scoprirlo, siamo passati ai nostri modelli murini di Alzheimer (i topolini APP23/PS45). Abbiamo provato due strategie:
1. Knockdown Genetico di ELK1: Abbiamo usato un virus adeno-associato (AAV) per “spegnere” il gene ELK1 specificamente nell’ippocampo, una regione chiave per la memoria. I risultati sono stati fantastici! Nei topi trattati, abbiamo osservato:
- Riduzione dei livelli di ELK1 e, di conseguenza, di PS1 (ma non di APP o BACE1).
- Diminuzione significativa dei livelli di Aβ40 e Aβ42.
- Riduzione del numero di placche senili nell’ippocampo.
- Miglioramento delle prestazioni nei test di memoria spaziale (Barnes maze e Morris water maze): i topi trovavano la via di fuga o la piattaforma nascosta più velocemente e con più precisione.
- Ripristino della plasticità sinaptica (misurata come potenziamento a lungo termine, LTP), un meccanismo cellulare fondamentale per l’apprendimento e la memoria, che era compromessa nei topi AD non trattati.
2. Inibizione Farmacologica di ELK1: Abbiamo usato un peptide chiamato TAT-DEF-ELK1 (TDE), che è progettato per bloccare specificamente la fosforilazione di ELK1 (cioè la sua attivazione). Lo abbiamo somministrato ai topi per via sistemica (iniezione intraperitoneale) per diversi mesi. Anche qui, i risultati sono stati estremamente incoraggianti e hanno rispecchiato quelli del knockdown genetico:
- Riduzione di PS1 nell’ippocampo.
- Diminuzione di Aβ e delle placche.
- Netto miglioramento delle capacità di apprendimento e memoria nei test comportamentali.
- Recupero della funzione sinaptica (LTP).
Importante sottolineare che il trattamento con TDE non sembrava causare effetti collaterali evidenti sui parametri generali di salute o sul comportamento ansioso/esplorativo dei topi, anche se ovviamente serviranno studi più approfonditi sulla sicurezza a lungo termine.

Cosa Significa Tutto Questo? Nuove Speranze per l’Alzheimer
Questa ricerca apre una strada davvero promettente. Abbiamo identificato un nuovo meccanismo attraverso cui l’Alzheimer progredisce: l’aumento di ELK1 (soprattutto p-ELK1) che, invece di agire come fattore di trascrizione in questo contesto, impedisce la normale degradazione di PS1 interferendo con SYVN1. Questo porta a un eccesso di PS1, maggiore attività della γ-secretasi, più Aβ, più placche e, alla fine, al declino cognitivo.
La cosa più entusiasmante è che abbiamo dimostrato che intervenire su questo meccanismo funziona! Sia bloccando ELK1 geneticamente sia inibendo la sua attivazione con un farmaco (il peptide TDE), siamo riusciti a ridurre la patologia amiloide e a migliorare significativamente la memoria e la funzione sinaptica nei modelli animali.
Questo suggerisce che ELK1 potrebbe essere un nuovo e valido bersaglio terapeutico per l’Alzheimer. Certo, la strada è ancora lunga. Bisognerà sviluppare magari piccole molecole o peptidi ancora più specifici, che blocchino l’interazione ELK1-PS1 senza interferire troppo con le altre funzioni fisiologiche di ELK1, e testarne la sicurezza e l’efficacia nell’uomo. Ma aver capito questo meccanismo e aver dimostrato che è “modulabile” con effetti benefici è un passo avanti importantissimo.
Insomma, la lotta contro l’Alzheimer continua, ma con una nuova freccia al nostro arco: puntare su ELK1 per ridare respiro alla memoria. Continuate a seguirci per futuri aggiornamenti!
Fonte: Springer





