Fibrosi Cutanea: Accendiamo i Motori Brucia-Grassi per Sconfiggerla?
Ragazzi, parliamoci chiaro: la fibrosi cutanea è una brutta bestia. Immaginate la vostra pelle che diventa progressivamente più spessa, rigida, quasi come un’armatura indesiderata. Questo è quello che succede in malattie come la Sclerosi Sistemica (SSc), e vi assicuro che non è solo un problema estetico. Limita i movimenti, può causare atrofia e, diciamocelo, peggiora drasticamente la qualità della vita. E la cosa frustrante? Le terapie veramente efficaci scarseggiano. Ecco perché quando inciampo in ricerche che aprono nuove strade, mi si accende una lampadina! Recentemente, l’attenzione si è spostata sul metabolismo, quel complesso insieme di reazioni chimiche che ci tiene in vita. E se la chiave fosse proprio lì, nascosta tra zuccheri, grassi e proteine?
Il Metabolismo Sotto la Lente: Cosa Succede nella SSc?
Abbiamo iniziato a indagare più a fondo, analizzando campioni di sangue di pazienti con SSc. E sapete cosa abbiamo trovato? Un’alterazione interessante nel metabolismo dei nucleotidi, in particolare tutto ciò che ruota attorno all’adenosina. I livelli di adenosina e dei suoi “parenti” (come ADP e AMP) erano significativamente più alti nei pazienti rispetto ai controlli sani. Ora, l’adenosina non è solo uno scarto metabolico; è una molecola segnale potentissima che agisce legandosi a specifici recettori sulle cellule, chiamati A1, A2A, A2B e A3. Questi recettori sono coinvolti in un sacco di processi: infiammazione, risposta immunitaria, persino la fibrosi stessa.
Il Recettore A2A: Un Protagonista Inatteso nei Fibroblasti
Ci siamo chiesti: quale di questi recettori gioca un ruolo chiave nella fibrosi cutanea, e soprattutto, cosa c’entrano i fibroblasti, le cellule operaie della nostra pelle responsabili della produzione di collagene (che nella fibrosi diventa eccessiva)? Abbiamo stimolato fibroblasti umani in laboratorio con varie citochine infiammatorie tipiche della SSc (come IL-1β, TGF-β, ecc.). Sorpresa: l’espressione del recettore A2A schizzava alle stelle, soprattutto con IL-1β! E andando a vedere direttamente nella pelle dei pazienti SSc, abbiamo confermato: i fibroblasti avevano molti più recettori A2A rispetto ai controlli sani. Bingo! Sembrava proprio che l’asse adenosina/A2A nei fibroblasti fosse un pezzo importante del puzzle.
Bloccare A2A: Una Mossa Vincente nei Modelli Animali
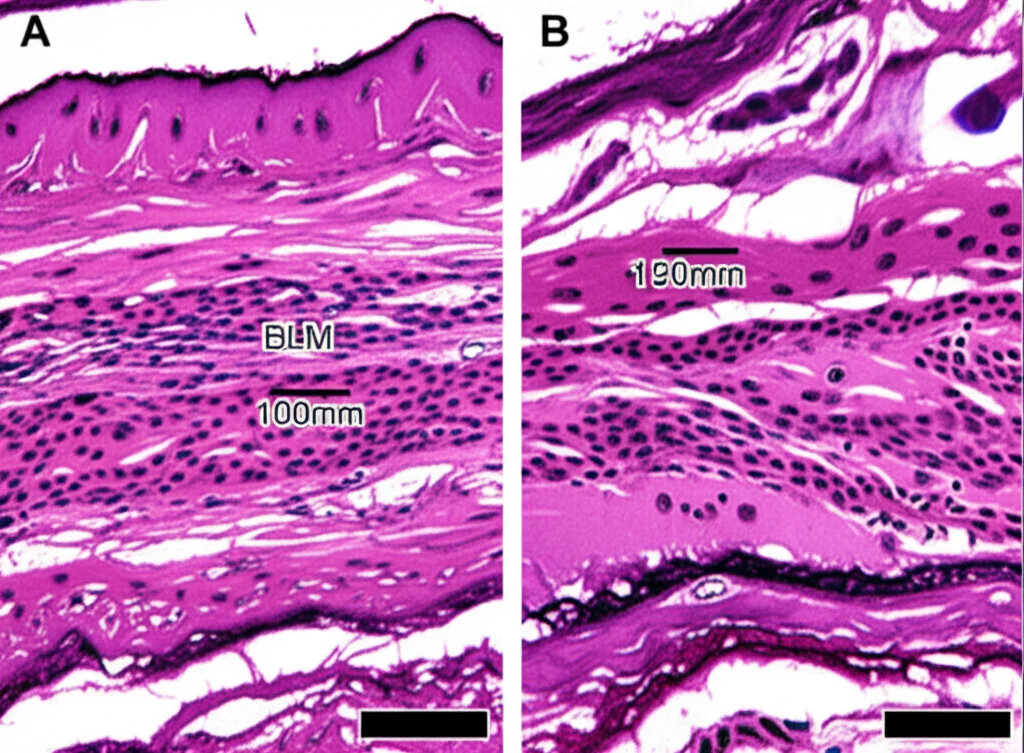
A questo punto, la domanda era ovvia: se blocchiamo questo recettore A2A, possiamo fermare o almeno rallentare la fibrosi? Per scoprirlo, abbiamo usato dei topolini geneticamente modificati per non avere il recettore A2A (knockout A2a). Abbiamo indotto la fibrosi cutanea in questi topolini (e in quelli normali, come controllo) usando due metodi diversi: iniezioni di bleomicina (BLM) e di acido ipocloroso (HOCL), entrambi noti per mimare la fibrosi della SSc. I risultati sono stati incredibili! Nei topolini senza A2A, la pelle si ispessiva molto meno, il deposito di collagene era ridotto, c’erano meno miofibroblasti (le cellule “cattive” della fibrosi) e anche i livelli dei geni associati alla fibrosi e all’infiammazione erano più bassi. Funzionava in entrambi i modelli! Questo ci ha dato una forte conferma: l’adenosina, agendo sul recettore A2A, promuove la fibrosi cutanea.
Dentro le Cellule: Sequenziamento a Singola Cellula Rivela un Indizio Metabolico
Per capire ancora meglio cosa succedeva *dentro* le cellule della pelle quando mancava A2A, abbiamo usato una tecnica potentissima: il sequenziamento dell’RNA a livello di singola cellula (scRNA-seq). Abbiamo analizzato migliaia di singole cellule dalla pelle dei nostri topolini (con e senza A2A, trattati e non trattati). Questo ci ha permesso di vedere quali geni erano accesi o spenti in ogni tipo di cellula (cheratinociti, fibroblasti, cellule immunitarie, ecc.). E indovinate un po’? Nei fibroblasti dei topi senza A2A, dopo l’induzione della fibrosi, abbiamo notato cambiamenti significativi nei geni legati al… metabolismo degli acidi grassi (FAO – Fatty Acid Oxidation)! Sembrava che bloccare A2A non solo fermasse i segnali pro-fibrotici classici, ma riaccendesse anche la capacità dei fibroblasti di “bruciare” i grassi. Una pista metabolica tutta nuova!
Fibroblasti in Provetta: A2A Spegne i Brucia-Grassi, Bloccandolo Li Riaccende!
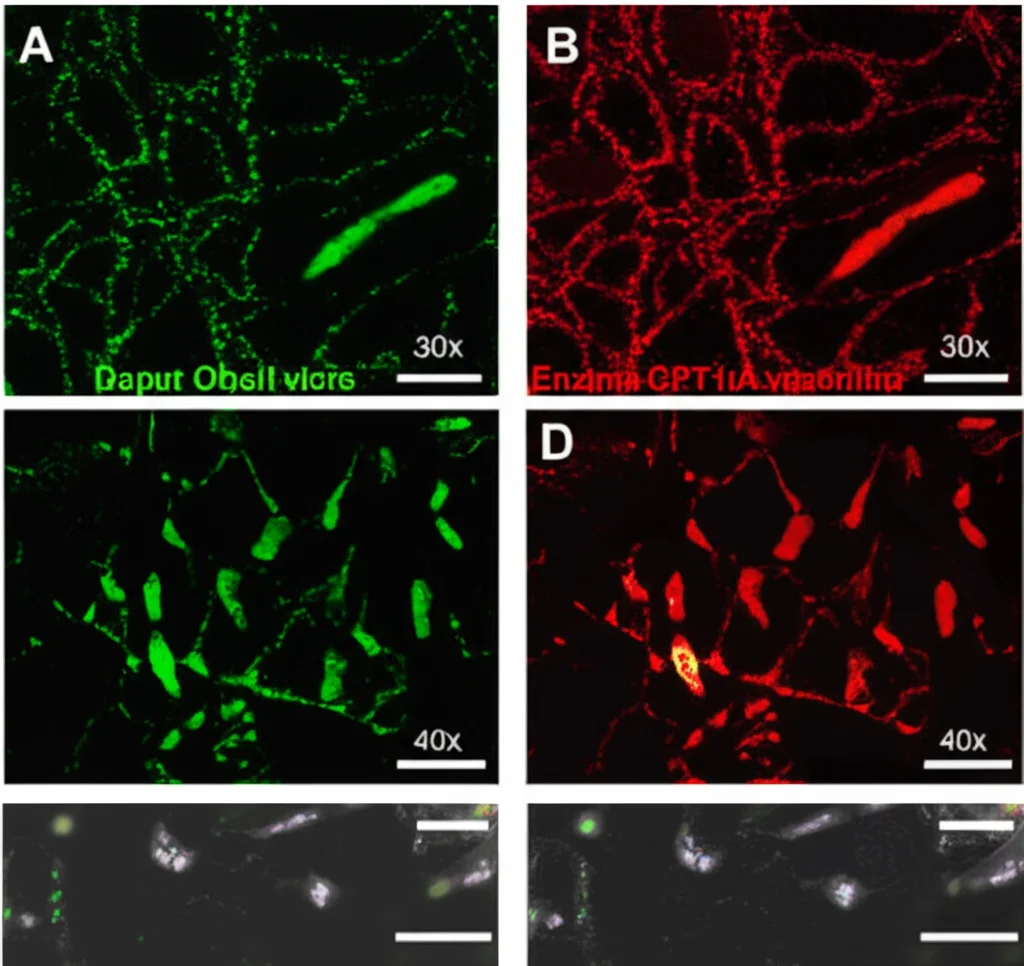
Tornati in laboratorio, abbiamo preso fibroblasti umani e li abbiamo trattati con TGF-β (un potente induttore di fibrosi) e con un inibitore specifico del recettore A2A (SCH442416) o un suo attivatore (CGS21680). Come previsto, l’inibitore A2A riduceva i segni della fibrosi (meno α-SMA, meno fibronectina, meno geni fibrotici attivi), mentre l’attivatore li peggiorava. Ma la vera chicca è arrivata dall’analisi dell’RNA (RNA-seq) di queste cellule. Il TGF-β, oltre a promuovere la fibrosi, spegneva letteralmente i geni della FAO. L’inibitore A2A, invece, faceva il contrario: contrastava l’effetto del TGF-β e riaccendeva i geni della FAO! In particolare, un gene chiave chiamato CPT1A, che è come l’interruttore principale per l’ossidazione degli acidi grassi, veniva spento dal TGF-β e riacceso dall’inibitore A2A.
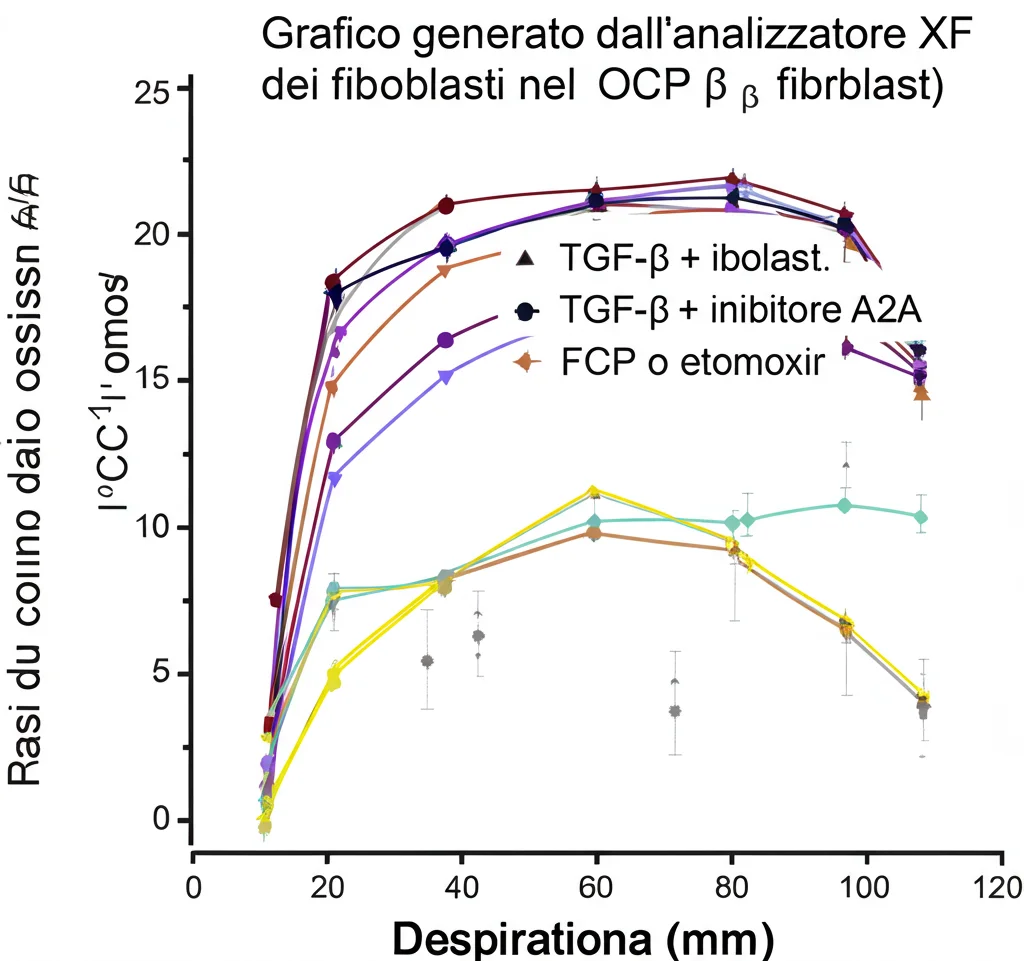
Abbiamo confermato tutto con un test chiamato Seahorse XF, che misura quanto “respirano” le cellule e quali carburanti usano. I risultati? I fibroblasti trattati con TGF-β usavano poco gli acidi grassi. Ma se aggiungevamo l’inibitore A2A, le cellule iniziavano a bruciare grassi a tutto spiano! Abbiamo anche misurato i prodotti di questo processo (acetilcarnitina e palmitoilcarnitina): bassi nei fibroblasti trattati con TGF-β, ma riportati alla normalità bloccando A2A. E, cosa importantissima, abbiamo ritrovato livelli bassi di questi metaboliti anche nel sangue dei pazienti SSc, confermando che questo problema della FAO non è solo in provetta, ma anche nella malattia reale. Per chiudere il cerchio, abbiamo usato un attivatore diretto della FAO (IVA337): da solo, riusciva a contrastare la fibrosi indotta da TGF-β e bloccava gli effetti pro-fibrotici dell’attivatore A2A. Insomma, sembra proprio che A2A promuova la fibrosi *spegnendo* la capacità dei fibroblasti di bruciare grassi.
Il Meccanismo Molecolare: Come A2A Controlla CPT1A?
Ok, A2A regola la FAO tramite CPT1A. Ma come fa esattamente? I recettori come A2A spesso funzionano attivando una cascata di segnali all’interno della cellula. Uno di questi segnali coinvolge una proteina chiamata CREB, che quando viene “fosforilata” (p-CREB) agisce come un interruttore genetico. Abbiamo visto che il TGF-β aumentava p-CREB nei fibroblasti, e l’inibitore A2A lo riduceva. Poteva essere questa la via? Abbiamo fatto altri esperimenti:
- Usando forskolina (che aumenta p-CREB), abbiamo visto che peggiorava la fibrosi e riduceva CPT1A, mimando l’effetto di A2A.
- Usando forskolina insieme all’inibitore A2A, abbiamo annullato l’effetto benefico dell’inibitore: la fibrosi tornava e CPT1A si spegneva di nuovo.
- Usando un inibitore specifico di p-CREB (666-15), abbiamo contrastato gli effetti negativi del TGF-β e dell’attivatore A2A: CPT1A si riaccendeva e la fibrosi diminuiva.
La sequenza sembrava chiara: Adenosina -> Attivazione A2A -> Aumento p-CREB -> Soppressione di CPT1A -> Riduzione della FAO -> Promozione della Fibrosi. Bloccando A2A, interrompiamo questa catena!
La Prova Finale: Knockout Specifico nei Fibroblasti
Per essere sicurissimi che fosse proprio l’A2A sui fibroblasti il colpevole, abbiamo creato dei topolini speciali in cui il gene A2a era eliminato *solo ed esclusivamente* nei fibroblasti (usando la tecnologia Cre-lox legata al gene Col1a2, specifico dei fibroblasti). Abbiamo ripetuto l’esperimento con la bleomicina. Risultato? Anche eliminando A2A solo nei fibroblasti, ottenevamo una protezione significativa dalla fibrosi cutanea: pelle meno spessa, meno collagene, meno miofibroblasti. E, come previsto dalla nostra ipotesi, in questi topi l’espressione di CPT1A nei fibroblasti della pelle era preservata nonostante l’insulto fibrotico. Abbiamo anche dato un’occhiata ai polmoni (altro organo colpito nella SSc) e abbiamo visto un miglioramento simile della fibrosi polmonare indotta da bleomicina. Questo conferma che colpire l’A2A specificamente nei fibroblasti è una strategia mirata ed efficace.
Cosa Ci Portiamo a Casa? Una Nuova Speranza Contro la Fibrosi
Quindi, ricapitolando: abbiamo scoperto che nella Sclerosi Sistemica c’è un eccesso di adenosina che, agendo sul recettore A2A presente sui fibroblasti, spegne un importante processo metabolico, l’ossidazione degli acidi grassi (FAO), attraverso la soppressione dell’enzima chiave CPT1A (via p-CREB). Questa alterazione metabolica contribuisce in modo decisivo alla fibrosi. La buona notizia? Bloccando il recettore A2A, possiamo riattivare la FAO e contrastare la fibrosi.
Questa scoperta è entusiasmante perché non solo ci fa capire meglio i meccanismi complessi dietro la fibrosi cutanea, ma apre la porta a nuove strategie terapeutiche. Immaginate farmaci che, invece di agire in modo generico, vadano a colpire specificamente l’asse A2A-FAO nei fibroblasti. Potrebbe essere un modo più mirato ed efficace per trattare la fibrosi cutanea nella SSc e forse anche in altre malattie fibrotiche. C’è ancora strada da fare, ovviamente, ma aver identificato questo legame cruciale tra metabolismo dell’adenosina e metabolismo dei grassi nella fibrosi è un passo avanti gigantesco. Incrociamo le dita!
Fonte: Springer





