
IA Svela i Segreti delle Proteine: Come le Modifiche Post-Traduzionali Influenzano i Farmaci
Ciao a tutti! Oggi voglio portarvi con me in un viaggio affascinante nel mondo microscopico delle nostre cellule, un luogo dove le proteine, i veri cavalli di battaglia della vita, non sono mai statiche. Immaginatele come operai super specializzati che, anche dopo essere stati “assunti” (cioè sintetizzati), ricevono ulteriori istruzioni e modifiche per svolgere al meglio il loro lavoro. Queste modifiche si chiamano Modifiche Post-Traduzionali, o PTM per gli amici.
Cosa sono le PTM e perché sono così importanti?
Le PTM sono come degli “upgrade” chimici che le cellule applicano alle loro proteine. Aggiungono piccoli gruppi chimici (fosfati, zuccheri, lipidi e altro) che possono cambiare radicalmente il comportamento di una proteina: la sua attività, stabilità, dove va all’interno della cellula e, cosa cruciale per noi oggi, come interagisce con altre molecole, inclusi i farmaci.
Pensateci: questa capacità di modificare le proteine al volo permette alle cellule di adattarsi rapidamente, rispondere a segnali e regolare finemente processi vitali come il metabolismo o la comunicazione cellulare. È un meccanismo incredibilmente elegante ed efficiente!
Purtroppo, quando qualcosa va storto con le PTM, le conseguenze possono essere serie. Difetti in queste modifiche sono collegati a un sacco di malattie: cancro, diabete, patologie cardiache, neurodegenerative e metaboliche. Capire le PTM è quindi fondamentale non solo per comprendere la biologia di base, ma anche per sviluppare nuove terapie.
La sfida: vedere l’invisibile (o quasi)
Uno degli aspetti più intriganti, e storicamente difficili da studiare su larga scala, è come le PTM influenzino la struttura tridimensionale delle proteine e, di conseguenza, come i farmaci si leghino ad esse. Una PTM può, ad esempio, cambiare la forma di una “tasca” sulla proteina dove un farmaco dovrebbe inserirsi, rendendo il farmaco meno efficace o addirittura inutile. Studiare questi dettagli strutturali richiedeva complessi e lenti metodi sperimentali. Ma qui entra in gioco la tecnologia!
L’Intelligenza Artificiale entra in scena
Negli ultimi anni, l’Intelligenza Artificiale (IA), in particolare gli algoritmi di deep learning, ha fatto passi da gigante nella previsione della struttura delle proteine. Strumenti come il famoso AlphaFold3, ma anche RoseTTAFold All-Atom (RFAA) e Chai-1, ci stanno aprendo porte che prima erano chiuse. Ora possiamo modellare al computer, con una precisione sorprendente e su larga scala, come appaiono le proteine, anche quelle modificate con PTM, e simulare come i farmaci interagiscono con esse. È una vera rivoluzione per la biologia strutturale computazionale!
Il nostro lavoro: mappare le PTM e i farmaci
Affascinati da queste nuove possibilità, ci siamo messi al lavoro. Abbiamo preso un database che avevamo sviluppato di recente, chiamato DrugDomain, che cataloga le interazioni tra domini proteici umani e piccole molecole (molti dei quali sono farmaci). Il nostro obiettivo era identificare quali PTM si trovassero proprio lì, vicino ai siti dove i farmaci si legano (abbiamo considerato una distanza di 10 Ångström, che è veramente un tiro di schioppo a livello molecolare).
Abbiamo scovato ben 6.131 PTM potenzialmente rilevanti per il legame dei farmaci! Per dare un contesto strutturale ed evolutivo, abbiamo anche mappato queste PTM sui domini proteici classificati secondo il database ECOD (Evolutionary Classification of Protein Domains).
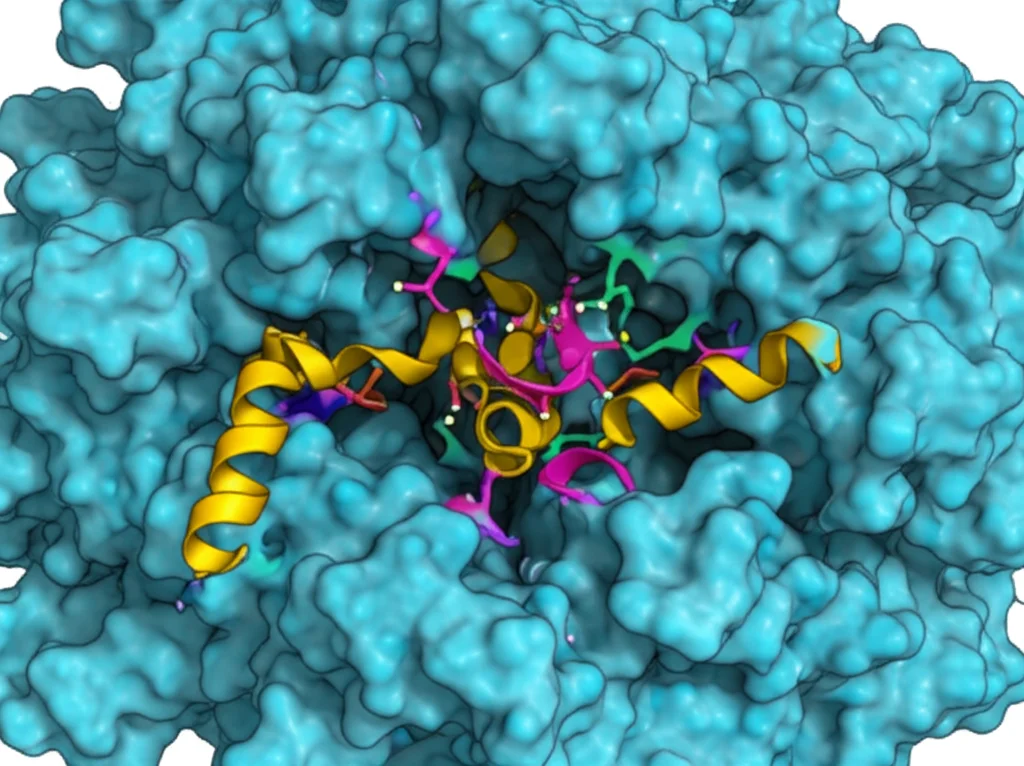
Quali PTM sono più comuni vicino ai siti di legame?
Analizzando i dati, abbiamo visto che tre tipi di PTM spiccavano per frequenza vicino ai siti di legame dei farmaci:
- Fosforilazione: L’aggiunta di un gruppo fosfato. È super comune perché è un interruttore on/off molto rapido per l’attività proteica, fondamentale nelle vie di segnalazione.
- Ubiquitinazione: L’aggiunta della proteina ubiquitina. Spesso segnala una proteina per la degradazione, ma ha anche molti altri ruoli regolatori.
- Acetilazione: L’aggiunta di un gruppo acetile. Cruciale, ad esempio, nella regolazione dell’espressione genica (modificando gli istoni) e nel metabolismo.
Abbiamo anche notato che queste PTM si trovavano spesso in domini proteici con architetture specifiche, come i “sandwich a tre strati a/b” (tipici degli enzimi metabolici come quelli con il fold di Rossmann), le topologie complesse a+b (dove troviamo molte chinasi, enzimi chiave nella segnalazione) e le strutture a “due strati a+b” (che includono proteine importanti come le heat shock proteins e domini come SH2 coinvolti nella regolazione di segnali).
Mettere alla prova l’IA: come se la cavano i modelli?
Ovviamente, non bastava identificare le PTM. Volevamo vedere se l’IA poteva davvero prevedere l’impatto strutturale di queste modifiche sul legame dei farmaci. Abbiamo usato AlphaFold3, RFAA, Chai-1 e un altro strumento chiamato KarmaDock per generare oltre 14.000 modelli di proteine umane modificate con PTM e con i rispettivi farmaci “agganciati” (docked).
Abbiamo iniziato testando questi metodi su un set di proteine dove era già noto o fortemente sospettato che una specifica fosforilazione influenzasse il legame di un farmaco. I risultati? Beh, sono stati… interessanti e un po’ contrastanti!
- RFAA e Chai-1: Questi strumenti sembravano in grado di catturare, in alcuni casi, l’effetto della PTM. A volte prevedevano correttamente che il farmaco si legasse in modo diverso o peggiore nella proteina modificata rispetto a quella non modificata. Tuttavia, Chai-1 mostrava a volte una certa incostistenza tra diverse simulazioni.
- AlphaFold3: Si è dimostrato molto bravo a prevedere la posizione del farmaco nella proteina *non* modificata, spesso con grande accuratezza rispetto alle strutture sperimentali. Però, nella maggior parte dei casi del nostro set di test, l’introduzione della PTM nel modello non cambiava significativamente la posizione prevista del farmaco. Sembrava meno “sensibile” all’effetto della modifica, almeno in questo contesto. C’è da dire, però, che in un caso specifico (il recettore IGF1), è stato l’unico a catturare correttamente uno stato conformazionale particolare della proteina.
- KarmaDock: Su questo set di test, non ha mostrato una grande accuratezza nel predire la posizione iniziale del farmaco.
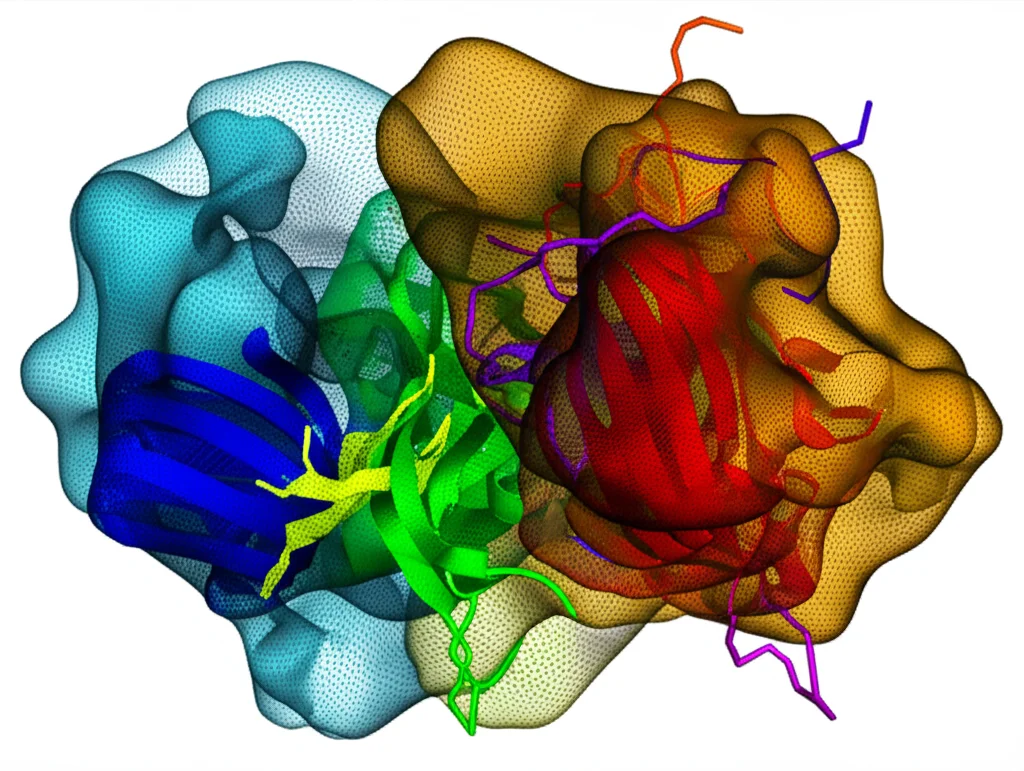
La conclusione di questa fase è che questi strumenti IA hanno un potenziale enorme, ma c’è ancora strada da fare. Probabilmente, la limitata quantità di dati sperimentali su strutture di proteine *con* PTM disponibili per addestrare questi modelli gioca un ruolo. Servirebbe un set di dati di benchmark molto più ampio per valutare davvero la loro precisione in questo compito specifico.
Una scoperta chiave: l’impatto della fosforilazione su un enzima legato al cancro
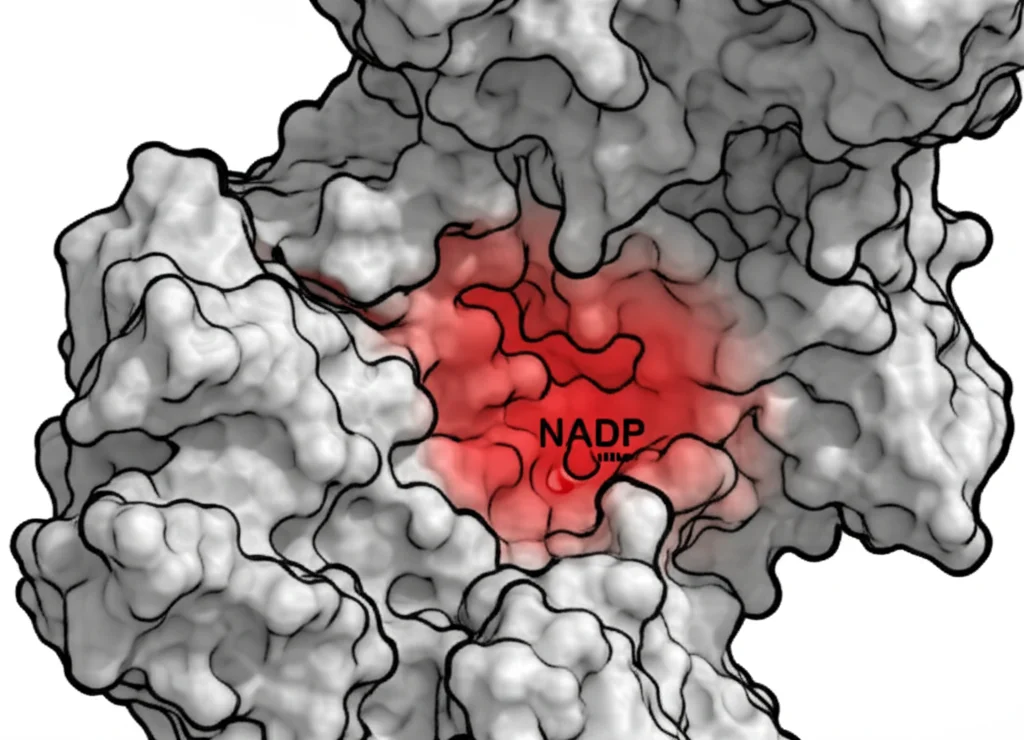
Nonostante le sfide, l’analisi dei nostri modelli ha portato a scoperte potenzialmente molto importanti. Un caso emblematico riguarda l’enzima NADPH-Citochromo P450 Reduttasi. Questo enzima è cruciale per il metabolismo di farmaci e sostanze estranee (xenobiotici) e per la produzione di steroidi.
Abbiamo scoperto che una specifica PTM, la fosforilazione di un residuo di tirosina (Tyr604), che era stata osservata in cellule di cancro della cervice e del polmone, sembra avere un impatto strutturale devastante. I nostri modelli, generati con tutti e quattro gli approcci (AlphaFold3, RFAA, Chai-1, KarmaDock), suggerivano che questa fosforilazione distorce significativamente la tasca di legame dove il substrato naturale dell’enzima, il NADP, dovrebbe alloggiarsi.
In particolare, i modelli di AlphaFold3 hanno mostrato che la tasca si restringe di circa 2 Ångström – una differenza piccola ma potenzialmente sufficiente a impedire al NADP di legarsi correttamente. Questo potrebbe compromettere seriamente la funzione dell’enzima. Non sorprende, quindi, che questa PTM sia stata trovata in cellule tumorali, dato che un malfunzionamento di questo enzima potrebbe influenzare processi critici per la carcinogenesi, come il metabolismo e lo stress ossidativo.
Una risorsa per tutti
La cosa bella della scienza moderna è la condivisione! Tutti i dati che abbiamo generato – l’elenco delle 6.131 PTM associate al legame di piccole molecole, la loro mappatura sui domini ECOD, e soprattutto i 14.178 modelli strutturali delle proteine modificate con i ligandi agganciati – li abbiamo resi pubblicamente disponibili.
Potete trovarli nella versione 1.1 del nostro database DrugDomain (http://prodata.swmed.edu/DrugDomain/) e su GitHub (https://github.com/kirmedvedev/DrugDomain). Per ogni proteina e ligando, c’è una tabella con le PTM identificate e link per visualizzare i modelli 3D (usando PyMOL). Crediamo che questa sia la prima risorsa su larga scala a fornire un contesto strutturale per le PTM rilevanti per il legame dei farmaci, e speriamo possa essere uno strumento prezioso per chiunque studi l’evoluzione, la struttura e la funzione delle proteine, e ovviamente per chi lavora nella scoperta di nuovi farmaci.
In conclusione
Siamo in un’epoca entusiasmante! L’IA ci sta dando strumenti potentissimi per esplorare la complessità della biologia a un livello di dettaglio prima impensabile. Il nostro lavoro ha mostrato come possiamo usare questi strumenti per iniziare a capire l’impatto strutturale delle PTM sul legame dei farmaci su scala proteomica. Abbiamo identificato migliaia di PTM potenzialmente importanti, valutato gli attuali strumenti IA (con i loro punti di forza e debolezza) e fatto scoperte specifiche, come quella sull’NADPH-Citochromo P450 Reduttasi, che potrebbero avere implicazioni cliniche.
Certo, c’è ancora molto da imparare e da migliorare, specialmente nel predire la flessibilità conformazionale delle proteine e l’effetto preciso di ogni PTM. Ma la strada è aperta, e non vedo l’ora di vedere cosa scopriremo dopo!
Fonte: Springer





