Helicobacter pylori e Cancro Gastrico: Come il “Cattivo” CagA Accende l’Infiammazione e Inganna le Nostre Difese
Ciao a tutti! Oggi voglio parlarvi di un argomento affascinante e, purtroppo, molto rilevante, specialmente qui in alcune parti del mondo: il cancro gastrico e il ruolo subdolo di un batterio molto comune, l’Helicobacter pylori. Sapete, circa metà della popolazione mondiale ospita questo microrganismo nel proprio stomaco, spesso senza saperlo. Ma per alcuni, questa convivenza non è pacifica e può portare a conseguenze serie, come gastriti croniche e, nei casi peggiori, al cancro dello stomaco.
Il problema è che, nonostante sappiamo da tempo che l’H. pylori è un fattore di rischio primario, capire *esattamente* come faccia a innescare il cancro è ancora un puzzle complesso. E questo rende difficile trovare metodi efficaci per la diagnosi precoce e terapie risolutive per gli stadi avanzati. L’immunoterapia, una speranza per molti tumori, funziona solo in una parte dei pazienti con cancro gastrico, e spesso si sviluppa resistenza. Perché? Una delle ipotesi più intriganti è che l’H. pylori stesso modifichi l’ambiente intorno al tumore, il cosiddetto microambiente tumorale (TME), influenzando come il cancro cresce e risponde alle cure.
Il Fattore di Virulenza CagA: L’Agente Provocatore
Al centro di questa storia c’è una proteina particolare prodotta da alcuni ceppi di H. pylori, chiamata CagA. Pensatela come un’arma segreta del batterio. I ceppi che producono CagA (CagA-positivi) sono considerati molto più pericolosi e associati a un rischio significativamente più alto di sviluppare il cancro gastrico. Ma come agisce questa proteina?
L’H. pylori usa un sofisticato sistema, simile a una siringa molecolare (il sistema di secrezione di tipo IV o T4SS), per iniettare CagA direttamente dentro le cellule della mucosa gastrica. Una volta dentro, CagA inizia a fare danni: interagisce con le vie di segnalazione della cellula ospite, mandandole in tilt. Questo porta a una serie di eventi negativi: le cellule perdono la loro normale organizzazione (polarità), iniziano a proliferare in modo incontrollato e subiscono trasformazioni che le avvicinano sempre di più allo stato maligno. È come se CagA riprogrammasse le cellule per farle diventare cancerose.
L’Inflammasoma NLRP3: Un Allarme Che Diventa Cronico
Ora entra in gioco un altro protagonista: l’inflammasoma NLRP3. Immaginatelo come un sistema di allarme molecolare all’interno delle nostre cellule. Normalmente, rileva segnali di pericolo (come infezioni batteriche o danni cellulari) e scatena una risposta infiammatoria per combattere la minaccia. Attiva il rilascio di potenti molecole infiammatorie, come le citochine IL-1β e IL-18, e può indurre un tipo particolare di morte cellulare infiammatoria chiamata piropotosi.
Tutto bene finché l’infiammazione è temporanea e mirata. Ma cosa succede se questo allarme rimane costantemente acceso? L’infiammazione cronica è un noto fattore che favorisce lo sviluppo dei tumori. E qui sta il punto: sembra che l’infezione da H. pylori, e in particolare la proteina CagA, sia in grado di attivare cronicamente l’inflammasoma NLRP3.
Nel nostro studio, analizzando dati da grandi database pubblici come TCGA e GEO, abbiamo visto che i livelli di mRNA di NLRP3 sono significativamente più alti nei tessuti tumorali gastrici rispetto a quelli normali, specialmente nei pazienti positivi all’H. pylori. L’analisi immunoistochimica (IHC) su campioni reali di pazienti ha confermato: c’è molta più proteina NLRP3 nei tumori gastrici associati all’infezione.

Il Meccanismo Svelato: CagA, miRNA e la Cascata Infiammaoria
Ma come fa esattamente CagA ad attivare NLRP3? Il percorso è affascinante e coinvolge anche piccole molecole regolatrici chiamate microRNA (miRNA). Abbiamo scoperto che CagA, una volta entrata nelle cellule gastriche (sia normali che cancerose), fa aumentare i livelli di un particolare microRNA, hsa-miR-1290.
Questo miR-1290, a sua volta, agisce come un interruttore molecolare, spegnendo un gene chiamato NKD1. Perché è importante? NKD1 normalmente agisce da freno per una via di segnalazione chiamata Wnt/β-catenina, che è spesso iperattiva nei tumori. E indovinate un po’? La via Wnt/β-catenina può attivare direttamente l’espressione di NLRP3!
Quindi, la catena di eventi sembra essere questa:
- L’H. pylori inietta CagA nella cellula gastrica.
- CagA fa aumentare i livelli di miR-1290.
- miR-1290 sopprime l’espressione di NKD1.
- La soppressione di NKD1 toglie il freno alla via Wnt/β-catenina.
- La via Wnt/β-catenina attivata promuove l’espressione dell’inflammasoma NLRP3.
Questa cascata porta non solo a più NLRP3, ma anche all’attivazione della piropotosi, con il rilascio di citochine infiammatorie (IL-1β, IL-18) e un aumento dello stress ossidativo (ROS) nelle cellule. Abbiamo confermato questi passaggi sia infettando cellule gastriche con H. pylori CagA-positivo, sia introducendo direttamente CagA nelle cellule tramite un plasmide. In entrambi i casi, abbiamo visto un aumento dell’apoptosi/piropotosi e dei marcatori infiammatori associati.
L’Impatto sul Microambiente Tumorale: Reclutare i “Guardiani Corrotti”
L’attivazione di NLRP3 non si limita a danneggiare le cellule tumorali stesse. Le citochine rilasciate (non solo IL-1β e IL-18, ma anche IL-6 e IL-10) si diffondono nel microambiente tumorale e influenzano il comportamento delle cellule immunitarie presenti. Qui le cose si fanno ancora più interessanti (e preoccupanti).
Abbiamo studiato in particolare l’effetto sui macrofagi, cellule immunitarie che possono avere un doppio ruolo nel cancro: possono essere “buoni” (M1), attaccando il tumore, o “cattivi” (M2), aiutando il tumore a crescere, a diffondersi e a sfuggire al sistema immunitario.
I nostri esperimenti *in vitro* hanno mostrato che le cellule di cancro gastrico che esprimono alti livelli di NLRP3 rilasciano più IL-6 e IL-10. Quando abbiamo esposto i macrofagi a queste citochine (o al “brodo di coltura” di queste cellule tumorali), abbiamo visto una chiara polarizzazione verso il fenotipo M2, quello pro-tumorale. I marcatori M2 (come CD206) aumentavano, mentre quelli M1 (come CD86) diminuivano.
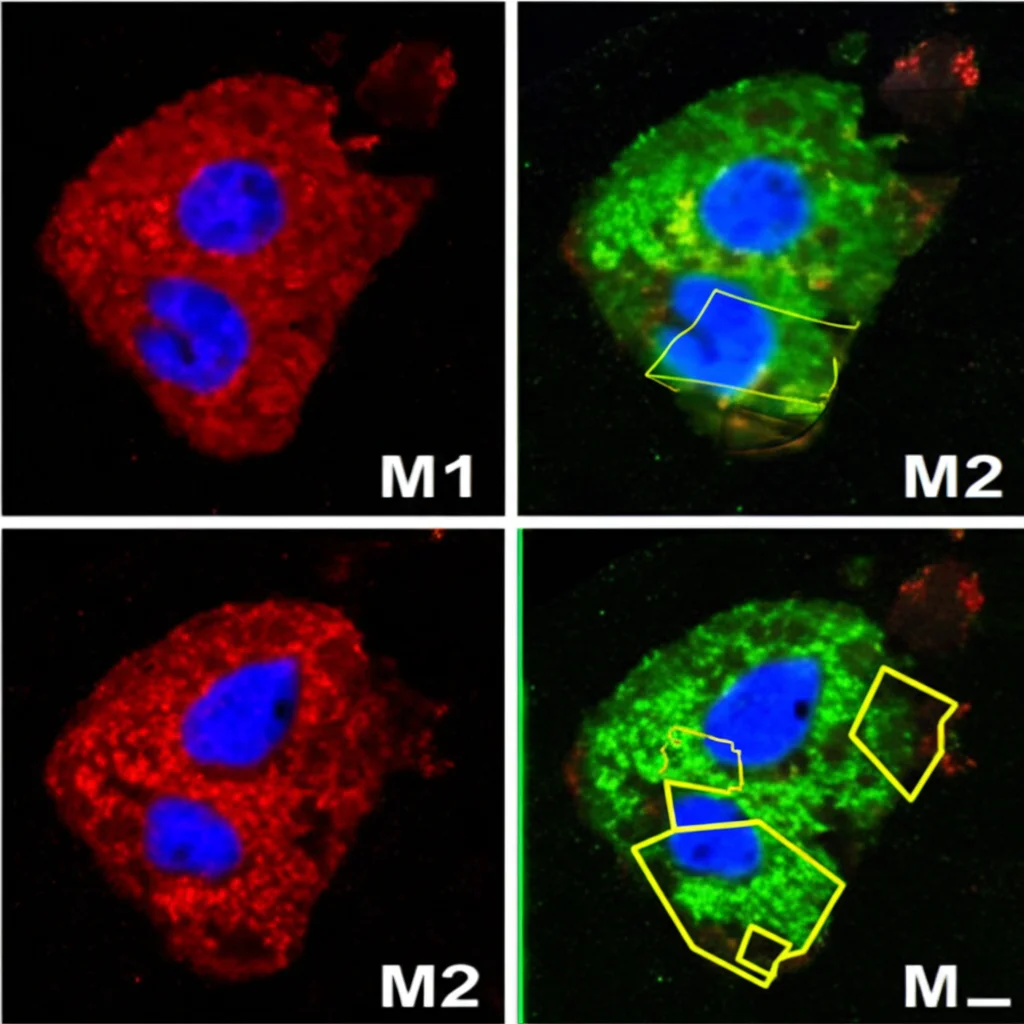
Come avviene questa polarizzazione? Sembra che IL-6 e IL-10 attivino una via di segnalazione specifica nei macrofagi, chiamata JAK2/STAT3. Abbiamo visto che i livelli di JAK2 e STAT3 fosforilati (cioè attivi) aumentavano nei macrofagi esposti alle citochine prodotte dalle cellule NLRP3-positive. Bloccando IL-6 e IL-10 con anticorpi neutralizzanti, riuscivamo a prevenire sia l’attivazione di JAK2/STAT3 sia la polarizzazione verso M2.
Quindi, NLRP3 nelle cellule tumorali orchestra indirettamente la creazione di un ambiente immunosoppressivo, ricco di macrofagi M2 “corrotti” che aiutano il tumore invece di combatterlo.
Un Panorama Immunitario Modificato e una Prognosi Peggiore
Questa manipolazione del sistema immunitario ha conseguenze concrete. Analizzando i dati di infiltrazione immunitaria nei tumori gastrici (usando strumenti bioinformatici come CIBERSORT e analizzando campioni reali con IHC), abbiamo trovato che alti livelli di NLRP3 sono associati a:
- Maggiore infiltrazione di macrofagi M2 (marcatore CD206).
- Maggiore infiltrazione di linfociti T regolatori (Tregs) e cellule T helper (CD4+). Queste cellule possono avere ruoli complessi, ma spesso contribuiscono a sopprimere la risposta anti-tumorale.
- Potenzialmente, una minore presenza di cellule T citotossiche (CD8+), i “soldati” principali contro il cancro.
In pratica, un’alta espressione di NLRP3 sembra rimodellare il campo di battaglia immunitario a favore del tumore.
E questo si riflette sulla prognosi dei pazienti. Abbiamo correlato i livelli di NLRP3 con le caratteristiche cliniche dei pazienti affetti da cancro gastrico. Risultato: alti livelli di NLRP3 sono associati a tumori più grandi, presenza di metastasi nei linfonodi, stadi TNM più avanzati (III-IV) e un grado di differenziazione peggiore (tumori meno simili al tessuto normale, quindi più aggressivi).
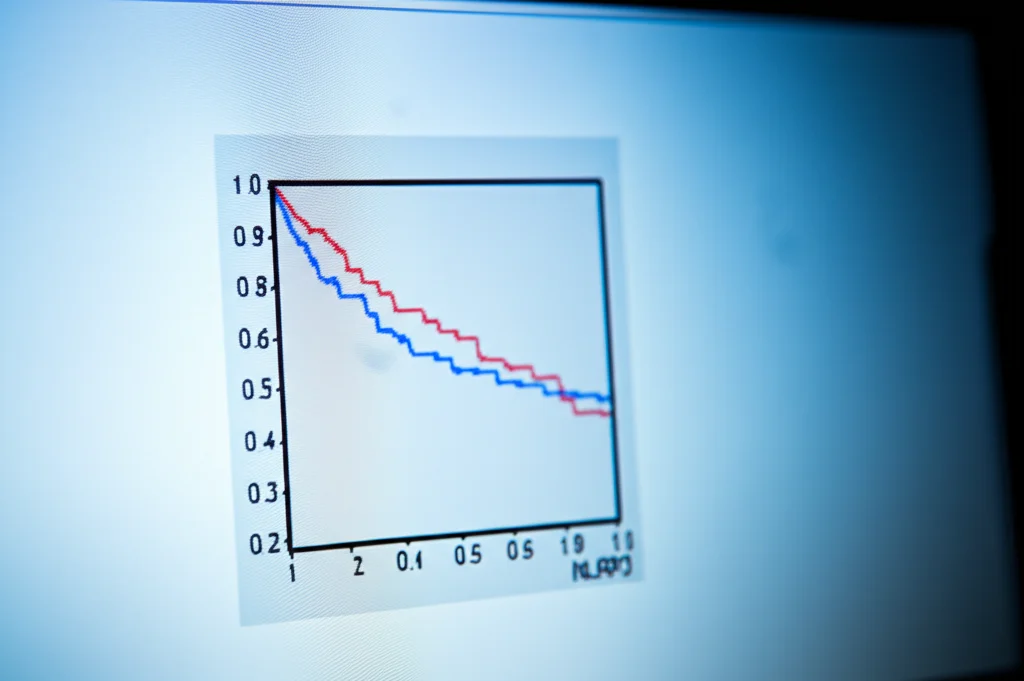
Infine, l’analisi della sopravvivenza è stata impietosa. Sia nei dati del database Kaplan-Meier Plotter che in quelli del TCGA, i pazienti con alti livelli di mRNA di NLRP3 nel tumore avevano una sopravvivenza globale (OS), una sopravvivenza libera da progressione (PFS) e una sopravvivenza post-progressione (PPS) significativamente peggiori rispetto ai pazienti con bassi livelli di NLRP3. Lo stesso valeva per i pazienti con alta infiltrazione delle cellule immunitarie “cattive” promosse da NLRP3, come le cellule CD4+ e CD206+ (M2).
Conclusioni e Prospettive Future: NLRP3 Come Bersaglio?
Mettendo insieme tutti i pezzi, emerge un quadro chiaro: l’infezione da H. pylori, tramite il suo fattore di virulenza CagA, innesca una cascata molecolare che porta all’iperattivazione dell’inflammasoma NLRP3 nelle cellule gastriche. Questo non solo promuove direttamente la crescita e l’invasività delle cellule tumorali attraverso la piropotosi e il rilascio di citochine, ma rimodella anche il microambiente tumorale, favorendo l’infiltrazione di cellule immunitarie soppressive come i macrofagi M2 e i Tregs. Il risultato finale è un tumore più aggressivo, capace di eludere il sistema immunitario, e una prognosi peggiore per il paziente.
Questa scoperta apre però nuove strade. Se NLRP3 è un attore così centrale nel guidare la progressione del cancro gastrico e nel creare un ambiente immunosoppressivo, allora bloccarlo potrebbe essere una strategia terapeutica promettente. Immaginate di poter “spegnere” questo interruttore infiammatorio per:
- Ridurre la crescita tumorale diretta.
- Diminuire il rilascio di citochine pro-tumorali come IL-6 e IL-10.
- Riprogrammare i macrofagi da M2 (cattivi) a M1 (buoni).
- Rendere il tumore più “visibile” e vulnerabile al sistema immunitario, magari aumentando l’efficacia dell’immunoterapia esistente.
Certo, la strada è ancora lunga e servono ulteriori ricerche per sviluppare farmaci specifici e sicuri che possano modulare l’attività di NLRP3 nel contesto del cancro gastrico. Ma aver identificato questo meccanismo è un passo avanti fondamentale. Ci aiuta a capire meglio la complessa danza tra infezione, infiammazione, immunità e cancro, e ci offre un nuovo potenziale bersaglio per combattere questa malattia devastante.
Fonte: Springer





