HPAP: Viaggio al Centro di un Enigmatico Tumore Cerebrale – Due Casi Che Fanno Luce
Avete presente quando vi trovate di fronte a un rompicapo, qualcosa che non si incasella facilmente nelle categorie che conoscete? Ecco, nel mio campo, quello della neuropatologia e dell’oncologia cerebrale, a volte ci imbattiamo in tumori che ci fanno grattare la testa. Uno di questi “nuovi arrivati” sulla scena, o meglio, uno di quelli che stiamo imparando a definire meglio, è il cosiddetto HPAP, acronimo che sta per High-grade glioma with pleomorphic and pseudopapillary features, ovvero glioma ad alto grado con caratteristiche pleomorfiche e pseudopapillari. Un nome un po’ lungo, lo so, ma che racchiude un mondo di complessità e, per fortuna, anche qualche speranza in più rispetto ad altri brutti ceffi come il glioblastoma IDH-wildtype.
Recentemente, insieme ai miei colleghi, abbiamo avuto l’opportunità di studiare da vicino due casi di HPAP che ci hanno insegnato parecchio e che, credo, possano aggiungere tasselli importanti alla comprensione di questa entità. E ve lo dico subito: quando si parla di tumori cerebrali, ogni nuova conoscenza è oro colato, perché può tradursi in diagnosi più precise e, si spera, in terapie più mirate.
Cos’è esattamente questo HPAP?
Prima di tuffarci nei nostri casi, cerchiamo di capire meglio chi è questo HPAP. Immaginate un gruppo di tumori cerebrali, i gliomi, che appaiono piuttosto ben circoscritti, ma che al microscopio mostrano un aspetto eterogeneo e spesso “arrabbiato”, cioè di alto grado. La cosa interessante è che, nonostante questa apparenza, i pazienti con HPAP sembrano avere una prognosi migliore rispetto a quelli con il temibile glioblastoma IDH-wildtype. Dal punto di vista genetico, questi tumori mostrano spesso alterazioni in geni come TP53, RB1, NF1, NF2 e BRAF. La loro “carta d’identità” molecolare, basata sull’analisi della metilazione del DNA, li raggruppa in un cluster specifico, distinto da altri tipi di glioma. Tuttavia, la domanda che ancora aleggia tra noi addetti ai lavori è: l’HPAP è davvero un’entità clinico-patologica a sé stante? I nostri casi, spero, contribuiranno a rispondere.
I nostri due “detective story” cliniche
Lasciate che vi presenti i protagonisti di questa storia, ovviamente in forma anonima per tutelare la loro privacy.
Il primo caso riguarda un ragazzo di 14 anni. Un’età così giovane per affrontare un problema tanto grande. Si è presentato con mal di testa forte, vomito e una paralisi facciale sinistra. La sua storia clinica era già complessa, con un ritardo nello sviluppo e perdita dell’udito. E, dettaglio non da poco, sulla pelle aveva delle macchie caffè-latte. La risonanza magnetica ha rivelato una grossa lesione nel cervello, tra i lobi parietale e occipitale destro, con una parte solida e una cistica, circondata da edema e che causava idrocefalo. C’erano anche altri noduli sospetti lungo i nervi cranici e spinali. Tutti questi indizi, messi insieme, hanno portato alla diagnosi di una condizione genetica chiamata Schwannomatosi correlata a NF2 (NF2-SCH), poi confermata da un test genetico. Il tumore principale è stato rimosso chirurgicamente e l’analisi ha suggerito proprio un HPAP. Dopo l’intervento, il ragazzo ha iniziato un percorso di chemio e radioterapia, e al momento, a 10 mesi dall’intervento, sta continuando la terapia di mantenimento con temozolomide.
Il secondo caso è quello di una giovane donna di 29 anni, che da tre mesi soffriva di un mal di testa sempre più insistente. Anche per lei, la risonanza magnetica ha mostrato una massa cerebrale, nei lobi parietale e frontale destro, con una componente solida e una grande cisti. Dopo l’asportazione chirurgica completa, la diagnosi è stata di glioma ad alto grado IDH-wildtype. Trattata con temozolomide, la paziente, a 42 mesi dall’intervento, sta bene e non ha segni di ripresa della malattia. Anche in questo caso, le caratteristiche istologiche e molecolari ci hanno indirizzato verso la famiglia degli HPAP.

Questi due pazienti, pur con storie diverse, ci hanno offerto una finestra privilegiata su questo tipo di tumore.
Sotto la lente: istologia e indizi molecolari
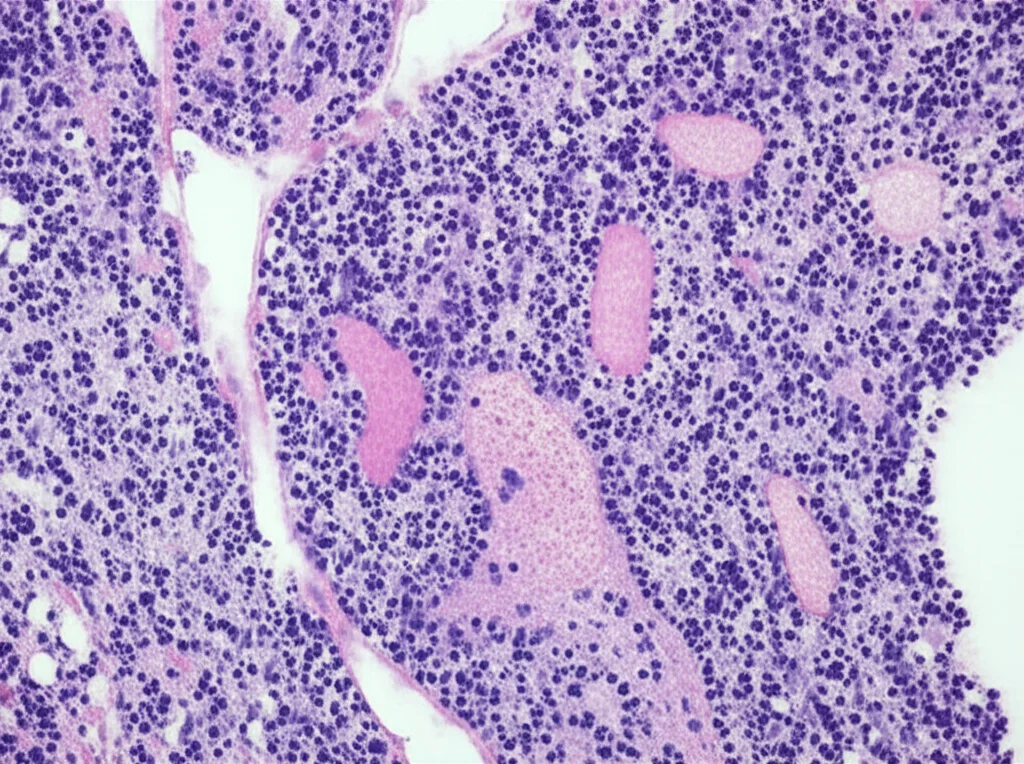
Quando abbiamo esaminato i campioni tumorali al microscopio, entrambi i casi mostravano tumori ben definiti rispetto al tessuto cerebrale circostante. Ma è qui che le cose si fanno affascinanti per un patologo!
- Nel caso 1, c’era una notevole eterogeneità: aree che ricordavano un ependimoma (un altro tipo di tumore cerebrale), altre con le tipiche strutture pseudopapillari, spesso separate da necrosi (morte cellulare), e zone con cellule marcatamente pleomorfiche, cioè di forme e dimensioni molto variabili e bizzarre. La maggior parte del tumore aveva caratteristiche di alto grado, ma, cosa importantissima, alla periferia c’era una componente di basso grado. L’espressione di alcune proteine (GFAP, CD34, p53) e la negatività per altre (OLIG2) hanno completato il quadro.
- Nel caso 2, il tumore aveva un aspetto che ricordava un astroblastoma, con cellule disposte attorno ai vasi a formare pseudorosette, intervallate da cellule giganti multinucleate. Anche qui, aree pseudopapillari, necrosi e mitosi (cellule in divisione). E, come nel primo caso, una forte e diffusa espressione della proteina p53, tranne che nella componente di basso grado del primo caso, che era p53-negativa.
L’analisi della metilazione del DNA, uno strumento potentissimo che ci aiuta a classificare i tumori cerebrali confrontandoli con un grande database (l’Heidelberg Brain Tumor Classifier), inizialmente ha dato un risultato di “no match” per entrambi i casi. Questo significa che non rientravano perfettamente in nessuna delle categorie note. Tuttavia, utilizzando un modello di classificazione preliminare, chiamato “Bethesda Classifier”, e un’analisi chiamata t-SNE, i nostri casi si sono collocati all’interno o molto vicino al gruppo HPAP. È come se l’Heidelberg fosse l’anagrafe ufficiale, e il Bethesda un investigatore più “di strada” che fiuta la pista giusta!
Dal punto di vista genetico, entrambi i tumori avevano varianti patogeniche nei geni RB1 e TP53. Nel primo caso, c’era anche una variante in NF2 (coerente con la sindrome NF2-SCH) e nel secondo una in CIC. La cosa davvero intrigante nel primo caso è che la componente di basso grado del tumore aveva solo la variante in NF2, suggerendo che il tumore sia evoluto da una lesione precursore a basso grado, guidata da NF2, che poi ha “fatto il salto” a alto grado acquisendo le alterazioni in RB1 e TP53. Una vera e propria evoluzione tumorale sotto i nostri occhi!
Perché questi casi sono importanti? Lezioni apprese
Vi chiederete: “Ok, interessante, ma cosa ce ne facciamo di tutte queste informazioni?”. Beh, parecchio!
- Localizzazione: Entrambi i nostri casi erano strettamente associati ai ventricoli laterali del cervello. Nello studio originale che ha descritto gli HPAP, solo un caso su 25 era intraventricolare. Questo suggerisce che l’HPAP dovrebbe essere preso in considerazione quando si diagnosticano lesioni vicino o dentro i ventricoli.
- Associazione con NF2-SCH: Il primo caso ci mostra una possibile associazione tra HPAP e la sindrome di Schwannomatosi correlata a NF2. Questo è un dato nuovo e importante, perché potrebbe spiegare l’insorgenza del tumore in età così giovane nel nostro paziente e, come detto, l’evoluzione da un precursore di basso grado. Sapere che pazienti con NF2-SCH potrebbero sviluppare questo tipo di tumore ci mette in allerta.
- Evoluzione da precursori di basso grado: L’osservazione di una componente di basso grado ben definita che evolve in una di alto grado è una novità per gli HPAP. Questo ci dice molto sulla biologia di questi tumori.
- Supporto all’HPAP come entità distinta: Ogni nuovo caso ben caratterizzato, come i nostri, aiuta a solidificare l’idea che l’HPAP sia un tipo di tumore con sue precise caratteristiche cliniche, istologiche e molecolari, e non solo una variante strana di qualcos’altro.
- Implicazioni terapeutiche: Riconoscere l’HPAP come un’entità con un comportamento potenzialmente intermedio, nonostante l’aspetto di alto grado, è cruciale. Potrebbe significare che non tutti i pazienti necessitano dei trattamenti aggressivi riservati, ad esempio, al glioblastoma. Nel nostro caso 1, abbiamo optato per radio e chemioterapia data l’aggressività istologica e le alterazioni genetiche, e similarmente per il caso 2 solo chemioterapia. Ma in futuro, con più dati, potremmo personalizzare ancora meglio le cure.

Insomma, il cammino della ricerca è fatto di piccoli passi, e questi due casi rappresentano un contributo in questa direzione. Ci dicono che dobbiamo tenere gli occhi aperti, continuare a studiare e integrare tutte le informazioni – cliniche, radiologiche, istologiche e molecolari – per dare un nome e un cognome precisi a questi tumori. E, soprattutto, per offrire ai nostri pazienti le migliori possibilità di cura.
Il mondo dei gliomi è in continua evoluzione, e l’HPAP è uno degli esempi più recenti di come la ricerca stia affinando sempre di più la nostra capacità di diagnosi. C’è ancora molta strada da fare, ma ogni scoperta ci avvicina un po’ di più all’obiettivo.
Fonte: Springer