FBL: La Proteina “Interruttore” che Accende il Cancro al Fegato (e Come Potremmo Spegnerla)
Ciao a tutti! Oggi voglio parlarvi di qualcosa di veramente affascinante che sta emergendo dalla ricerca sul cancro, in particolare su quello che colpisce il fegato, l’epatocarcinoma (HCC). Sapete, l’HCC è un osso duro, una delle principali cause di morte per cancro nel mondo, e nonostante i progressi, abbiamo un disperato bisogno di nuove armi per combatterlo.
Per anni, farmaci come il sorafenib sono stati lo standard, ma diciamocelo, i risultati non erano entusiasmanti. Poi sono arrivate le terapie combinate, anche con l’immunoterapia, che hanno migliorato le cose. Ma il problema delle recidive rimane, e molti pazienti si ritrovano con un tumore avanzato e incurabile. Ecco perché la ricerca di nuovi bersagli terapeutici è fondamentale. E non solo: servono anche biomarcatori più efficaci per la diagnosi precoce, perché l’attuale alfa-fetoproteina (AFP) non è sempre affidabile, specialmente nelle fasi iniziali.
L’Epigenetica: Registi Nascosti del Destino Cellulare
Qui entra in gioco un campo affascinante: l’epigenetica. Immaginatela come un insieme di “interruttori” e “manopole” che controllano quali geni vengono accesi o spenti nelle nostre cellule, senza cambiare il DNA stesso. È sempre più chiaro che quando questi controlli impazziscono (disregolazione epigenetica), possono contribuire all’insorgenza e alla progressione dei tumori, incluso l’HCC.
Si sta lavorando molto sui cosiddetti “epidrugs”, farmaci che mirano a correggere questi problemi epigenetici. Alcuni sono già approvati per tumori del sangue e testati per tumori solidi come l’HCC. Tuttavia, la strada è ancora lunga, soprattutto per questioni di sicurezza. Capire meglio queste alterazioni epigenetiche è cruciale per trovare nuovi bersagli, migliorare la diagnosi e aumentare l’efficacia delle terapie.
La Scoperta di FBL: Un Protagonista Inatteso
Nel nostro lavoro, abbiamo deciso di scandagliare i dati genetici di pazienti con HCC (usando un database pubblico chiamato TCGA) per cercare proprio quei geni epigenetici che fossero “fuori controllo”. Analizzando decine di questi geni, uno in particolare ha attirato la nostra attenzione per quanto fosse espresso in modo anomalo: FBL (Fibrillarina).
FBL non è un nome nuovo in biologia. È un enzima (una rRNA 2′-O-metiltransferasi) noto per il suo ruolo nella “maturazione” dell’RNA ribosomiale e per modificare un particolare tipo di proteina istonica (H2A). Studi precedenti l’avevano già collegato ad altri tumori (seno, pancreas, esofago), associandolo a una prognosi peggiore. Si sapeva anche che FBL può influenzare la produzione di proteine chiave per la crescita tumorale. Ma il suo ruolo specifico nell’HCC e i meccanismi precisi erano ancora avvolti nel mistero.
Abbiamo quindi verificato: i livelli della proteina FBL erano davvero molto più alti nei tessuti tumorali di HCC rispetto ai tessuti sani adiacenti, nella stragrande maggioranza dei pazienti analizzati (oltre l’80%!). E, cosa importante, i pazienti con livelli più alti di FBL avevano una sopravvivenza più bassa e spesso un tumore in stadio più avanzato. Tutti questi indizi puntavano nella stessa direzione: FBL sembrava comportarsi da oncogene, un gene che promuove il cancro.
FBL in Azione: Il Motore della Crescita Tumorale
Ma non ci siamo fermati agli indizi. Volevamo vedere FBL “in azione”. Abbiamo preso delle cellule di HCC in laboratorio (quelle che esprimevano più FBL) e abbiamo usato delle tecniche per “spegnere” il gene FBL (knockdown). Il risultato? Le cellule tumorali hanno iniziato a proliferare molto meno e a formare meno colonie (un test che simula la capacità di formare un tumore). Al contrario, quando abbiamo “acceso” di più FBL in altre cellule tumorali (sovraespressione), queste hanno iniziato a crescere più velocemente.
Poi siamo passati agli esperimenti in vivo, usando modelli animali. Abbiamo iniettato le cellule con FBL “spento” in topi speciali (immunodeficienti) e abbiamo visto che i tumori crescevano molto più lentamente e rimanevano più piccoli rispetto a quelli formati dalle cellule di controllo. Questi risultati confermavano che FBL è davvero un motore importante per la crescita dell’HCC.
Chi Accende FBL? La Pista Porta a RBPJ
Ok, FBL è importante. Ma cosa lo fa esprimere così tanto nel tumore? Analizzando altri database e correlando l’espressione dei geni, abbiamo trovato un forte candidato come “regista” di FBL: un fattore di trascrizione chiamato RBPJ. Abbiamo verificato che spegnendo RBPJ nelle cellule tumorali, anche i livelli di FBL diminuivano significativamente. Esperimenti più specifici (come i saggi con reporter luciferasi e ChIP) hanno confermato che RBPJ si lega direttamente al “promotore” del gene FBL (la sua sequenza di accensione) e ne aumenta l’espressione.
Interessante notare che anche RBPJ stesso è risultato più espresso nei tumori HCC e associato a una prognosi peggiore, suggerendo che agisca anch’esso come un oncogene, almeno in parte proprio accendendo FBL. Abbiamo anche verificato che questa regolazione da parte di RBPJ avviene indipendentemente da un’altra via di segnalazione cellulare nota (la via di NOTCH).
Svelare la Catena di Comando: FBL, YY1 e CAD
A questo punto, la domanda era: come fa FBL a promuovere la crescita tumorale una volta che è acceso? Quali sono i suoi “soldati” a valle? Abbiamo cercato geni la cui espressione seguisse quella di FBL e ne abbiamo identificati alcuni candidati. Tra questi, uno spiccava per la sua forte correlazione con la sopravvivenza dei pazienti: il gene CAD.
CAD è un nome importante nel metabolismo cellulare. È un enzima multifunzionale cruciale per la sintesi delle pirimidine, mattoncini fondamentali per costruire il DNA e l’RNA. Senza abbastanza pirimidine, le cellule non possono duplicarsi rapidamente, come fanno quelle tumorali.
Abbiamo verificato: spegnendo FBL, diminuivano anche i livelli di CAD (sia mRNA che proteina). Inoltre, spegnendo FBL o CAD, le cellule tumorali mostravano un blocco nel ciclo cellulare (in fase G1) e una ridotta sintesi di DNA, proprio come ci si aspetterebbe se mancassero i mattoncini per costruirlo. Anche spegnere direttamente CAD riduceva la proliferazione cellulare e la crescita tumorale nei topi. E, guarda caso, anche CAD era più espresso nei tumori HCC e correlato a una prognosi sfavorevole. Sembrava proprio che FBL agisse tramite CAD. Ma come?
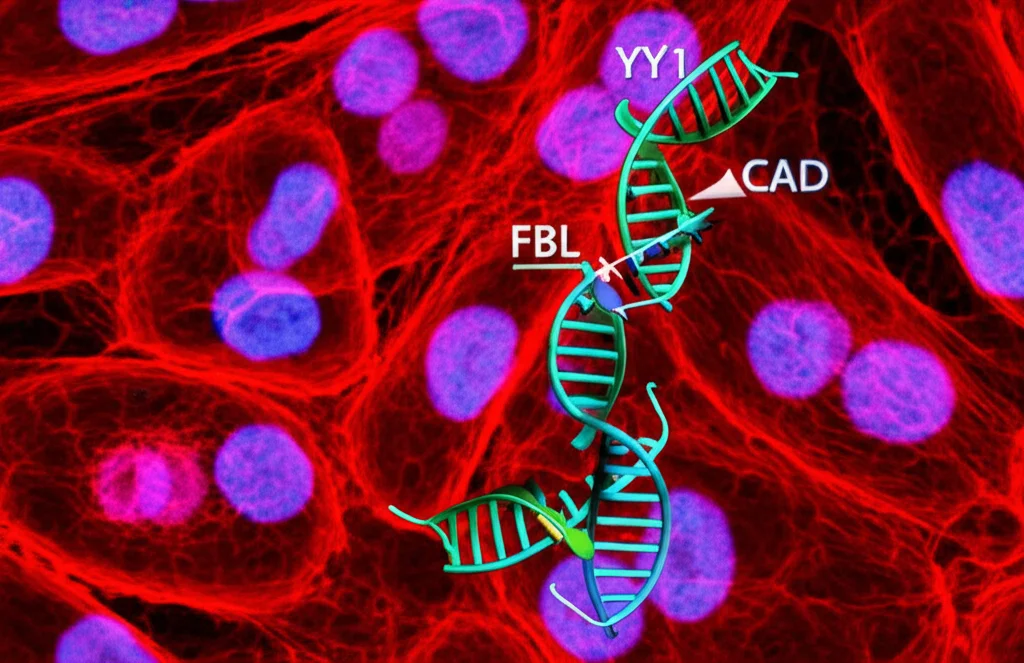
Qui la storia si fa ancora più intrigante. Abbiamo cercato proteine che interagissero direttamente con FBL e abbiamo trovato un altro fattore di trascrizione: YY1 (Yin Yang 1). Esperimenti di immunoprecipitazione hanno confermato che FBL e YY1 si legano fisicamente dentro le cellule tumorali e si trovano spesso insieme (colocalizzazione).
YY1 è un fattore “versatile”, coinvolto in molti processi cellulari. Abbiamo scoperto che anche YY1 regola l’espressione di CAD: spegnendo YY1, i livelli di CAD diminuivano, mentre accendendolo aumentavano. Inoltre, YY1 si lega al promotore di CAD.
La cosa sorprendente è che FBL non sembrava regolare i livelli di YY1. Allora, come funziona? L’ipotesi più logica era che FBL e YY1 collaborassero.
Un Meccanismo Inaspettato: FBL come “Reclutatore”
Pensavamo inizialmente che FBL, essendo un enzima che modifica gli istoni (un “writer” epigenetico), potesse regolare CAD modificando la cromatina (il pacchetto di DNA e proteine) vicino al gene. Ma le analisi (ATAC-seq, CUTeRUN) hanno mostrato che FBL non cambiava l’accessibilità del promotore di CAD né aggiungeva il suo “marchio” tipico (la metilazione H2AQ104) in quella zona.
Allora abbiamo guardato più da vicino la struttura della proteina FBL. Usando strumenti bioinformatici, abbiamo identificato una regione nascosta in FBL (tra gli amminoacidi 206-274) che assomiglia a un “dominio di transattivazione” (TAD), una specie di “calamita” molecolare capace di reclutare altri fattori per attivare i geni. Esperimenti successivi hanno confermato che questo dominio TAD di FBL è capace di attivare la trascrizione e che interagisce proprio con YY1.
Ecco il quadro che emerge: FBL, attraverso il suo dominio TAD, recluta il fattore di trascrizione YY1 sul promotore del gene CAD, potenziandone l’espressione. È come se FBL dicesse a YY1: “Ehi, vieni qui e accendi questo gene CAD!”. Questo meccanismo è indipendente dall’attività enzimatica “classica” di FBL come metiltransferasi. Infatti, anche versioni mutate di FBL, incapaci di svolgere la loro funzione enzimatica, erano ancora in grado di promuovere la proliferazione cellulare!

Spegnere l’Interruttore: Fludarabina Fosfato come Inibitore di FBL
Se FBL è così importante per la crescita dell’HCC, bloccarlo potrebbe essere una strategia terapeutica vincente. Abbiamo quindi cercato molecole capaci di inibire FBL. Analizzando virtualmente milioni di composti e testandone alcuni in laboratorio, abbiamo identificato un candidato promettente: la Fludarabina Fosfato.
Questo nome potrebbe suonare familiare ad alcuni, perché è un farmaco già approvato dalla FDA e usato per trattare leucemie e linfomi. Abbiamo scoperto, per la prima volta, che la Fludarabina può legarsi direttamente a FBL (confermato da saggi termici cellulari, pull-down e SPR). Simulazioni al computer (docking) suggeriscono che potrebbe legarsi proprio vicino al dominio TAD, interferendo forse con l’interazione con YY1.
E infatti, trattando le cellule tumorali con Fludarabina, abbiamo visto che l’attività trascrizionale di YY1 diminuiva e, di conseguenza, calavano anche i livelli di CAD. La Fludarabina riusciva a inibire la proliferazione delle cellule HCC in modo dipendente dalla dose e dal tempo. È importante notare che la Fludarabina non sembrava bloccare l’attività enzimatica “classica” di FBL (metilazione di H2AQ104 o sintesi pre-rRNA), rafforzando l’idea che agisca sul suo ruolo di “reclutatore” tramite il dominio TAD.
Un Duo Potente: Fludarabina + Lenvatinib
Oggi, nel trattamento dei tumori, si punta molto sulle terapie combinate per aumentare l’efficacia e ridurre le resistenze. Uno dei farmaci usati per l’HCC avanzato è il Lenvatinib, un inibitore multichinasico. Ci siamo chiesti: cosa succederebbe combinando Fludarabina e Lenvatinib?
I risultati sono stati entusiasmanti! In laboratorio, la combinazione dei due farmaci ha mostrato un effetto sinergico: l’effetto antitumorale era maggiore della somma degli effetti dei singoli farmaci. Abbiamo visto che anche Lenvatinib, per conto suo, riduceva un po’ l’espressione di CAD, probabilmente attraverso le vie di segnalazione che blocca (VEGFR, FGFR etc.). La combinazione dei due farmaci portava a una riduzione ancora più marcata dei livelli di CAD.
Siamo quindi passati ai modelli animali. Nei topi con tumori HCC derivati da cellule umane (CDX), la Fludarabina da sola riduceva la crescita tumorale, ma la combinazione con Lenvatinib la bloccava quasi completamente, anche a dosi basse di Fludarabina. Risultati simili li abbiamo ottenuti in un modello ancora più realistico, usando tumori prelevati direttamente da pazienti e trapiantati nei topi (PDX). Anche qui, la combinazione Fludarabina + Lenvatinib è risultata significativamente più efficace dei singoli trattamenti nel ridurre le dimensioni del tumore e i livelli della proteina CAD al suo interno.

Conclusioni e Prospettive Future
Quindi, cosa abbiamo imparato? Abbiamo identificato un nuovo asse molecolare cruciale per la crescita del cancro al fegato: FBL-YY1-CAD. Abbiamo scoperto che FBL, una proteina sovraespressa nell’HCC e legata a una prognosi peggiore, non agisce solo tramite la sua nota funzione enzimatica, ma possiede un dominio “nascosto” (TAD) che le permette di reclutare il fattore YY1 sul promotore del gene CAD. Questo potenzia la produzione di CAD, un enzima chiave per la sintesi delle pirimidine, fornendo così il “carburante” necessario alle cellule tumorali per proliferare.
La scoperta più entusiasmante, forse, è che un farmaco già esistente, la Fludarabina Fosfato, può inibire FBL, probabilmente interferendo con questa funzione di reclutamento, e bloccare la crescita dell’HCC. Ancora meglio, la Fludarabina agisce in sinergia con il Lenvatinib, un farmaco già in uso clinico per l’HCC.
Questo apre nuove, concrete speranze terapeutiche. La combinazione Fludarabina + Lenvatinib potrebbe rappresentare una strategia più efficace per trattare pazienti con HCC, magari superando anche le resistenze alle terapie attuali. Ovviamente, siamo ancora in una fase preclinica, ma i risultati sono molto promettenti.
Il nostro lavoro sottolinea l’importanza di scavare a fondo nei meccanismi molecolari del cancro e di come l’epigenetica offra bersagli inaspettati. Ci sono ancora domande aperte: FBL e YY1 regolano altri geni insieme? Quali sono i biomarcatori migliori per predire la risposta a questa potenziale nuova terapia? La ricerca continua, ma abbiamo aggiunto un tassello importante alla comprensione e, speriamo, al futuro trattamento di questa grave malattia.
Fonte: Springer