
Viaggio nel Cuore del Riso: L’Evoluzione Svelata dai Genomi Tetraploidi
Ciao a tutti! Oggi voglio portarvi con me in un viaggio affascinante nel mondo del riso, o meglio, nel genere Oryza, la grande famiglia a cui appartiene il riso che tutti conosciamo. Sapete, il riso coltivato (Oryza sativa e Oryza glaberrima) è fondamentale per l’alimentazione di miliardi di persone, ma millenni di domesticazione hanno creato un “collo di bottiglia genetico”. In pratica, abbiamo selezionato le caratteristiche migliori per noi, ma abbiamo perso un sacco di diversità genetica preziosa che si trova ancora nei suoi parenti selvatici.
Questa diversità è una vera miniera d’oro! Contiene geni che potrebbero aiutarci a creare varietà di riso più resistenti alla siccità, alle malattie, alla salinità, insomma, più adatte alle sfide del cambiamento climatico e a sfamare una popolazione mondiale in crescita. Per questo, ho avuto la fortuna di partecipare a uno studio incredibile: abbiamo deciso di “aprire la cassaforte” genetica del genere Oryza.
Un Puzzle Genetico Complesso: Il Genere Oryza
Il genere Oryza è una meraviglia della natura: comprende 27 specie, tra cui 25 selvatiche, suddivise in 11 tipi di genomi diversi (indicati con sigle come AA, BB, CC, BBCC, CCDD, ecc.). Alcune specie sono diploidi (come noi, con due copie di ogni cromosoma, 2n=2x=24), altre sono allotetraploidi (nate dall’incrocio di due specie diverse, con quattro copie dei cromosomi, 2n=4x=48). Pensate che la dimensione del loro genoma può variare tantissimo, quasi 3.4 volte tra la specie più piccola e quella più grande! Questa famiglia si è evoluta nel corso di circa 15 milioni di anni, accumulando un patrimonio genetico immenso e in gran parte inesplorato.
Il nostro obiettivo? Sequenziare e analizzare a fondo i genomi di specie finora poco studiate, soprattutto quelle tetraploidi. Abbiamo generato 11 nuovi genomi di riferimento a livello cromosomico (cioè mappe genetiche super dettagliate) per 9 specie tetraploidi selvatiche e 2 diploidi selvatiche, usando tecnologie all’avanguardia come il sequenziamento PacBio a lettura lunga e la validazione con mappe ottiche Bionano. Un lavoro enorme, ma ne è valsa la pena!
Il Cuore Conservato e la Periferia Dinamica
Mettendo insieme i nostri 11 nuovi genomi con quelli già disponibili, abbiamo potuto osservare l’evoluzione del genoma Oryza con una lente d’ingrandimento senza precedenti. Cosa abbiamo scoperto? Che esiste un “cuore” del genoma, una porzione di circa 200 Megabasi (Mb), che è rimasta sorprendentemente simile e ordinata (i genetisti dicono “sintenica”) in quasi tutte le specie, nonostante milioni di anni di evoluzione separata. È come se ci fosse uno scheletro fondamentale comune a tutta la famiglia.
Tutto il resto del genoma, invece, che può variare da 80 a 600 Mb a seconda della specie, è molto più “plastico”, dinamico e in rapida evoluzione. E chi sono i principali architetti di questa variabilità? Gli elementi trasponibili (TE), noti anche come “geni saltellanti”. Questi frammenti di DNA possono copiarsi e incollarsi in diverse parti del genoma, facendolo espandere o contrarre.
Abbiamo visto un esempio lampante nel cosiddetto “complesso ridleyi” (specie con genoma HHJJ come O. ridleyi e O. longiglumis). Qui, i genomi sono diventati enormi (oltre 1100 Mb!). La cosa interessante è che uno dei due sub-genomi ancestrali (l’HH) è cresciuto molto più dell’altro (il JJ), circa 1.5 volte! Analizzando i TE, abbiamo capito che questa differenza non è dovuta all’esplosione di poche famiglie di TE specifiche, né a una diversa efficienza nel rimuoverli, ma piuttosto a un’accumulazione preferenziale di tanti tipi diversi di TE nel sub-genoma HH dopo l’evento di poliploidizzazione (l’unione dei due genomi ancestrali). Insomma, un sub-genoma è diventato “più avido” di TE rispetto all’altro.

Quando i Cromosomi si Rimescolano: La Macrosintenia
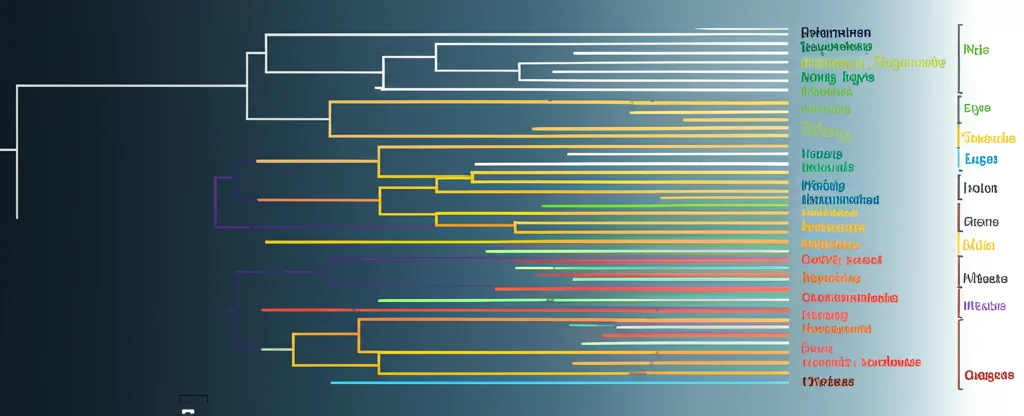
Guardando i genomi su larga scala, abbiamo costruito una mappa di “sintenia” (conservazione dell’ordine dei geni sui cromosomi) per l’intero genere Oryza, includendo 21 specie e persino un parente più lontano (Leersia perrieri) come confronto. È come avere una mappa stradale che mostra come le “autostrade” cromosomiche si sono mantenute, interrotte o scambiate di posto nel corso di 15 milioni di anni.
Abbiamo visto grandi inversioni, duplicazioni e traslocazioni (pezzi di cromosoma che si spostano su un altro cromosoma). Un caso interessante riguarda Oryza alta e Oryza grandiglumis, entrambe con genoma CCDD. Condividono ben cinque grandi traslocazioni sbilanciate rispetto al riso coltivato (AA) e anche rispetto a un’altra specie CCDD, Oryza latifolia. Questo è un forte indizio molecolare che supporta l’ipotesi, già avanzata su basi morfologiche e citogenetiche, che O. alta e O. grandiglumis siano in realtà la stessa specie o molto, molto vicine tra loro (conspecifiche). Curiosamente, abbiamo notato che vicino ai punti di rottura di queste traslocazioni ci sono spesso sequenze di DNA molto semplici, come ripetizioni di ATATAT… Forse queste sequenze hanno reso i cromosomi più “fragili” in quei punti, facilitando i riarrangiamenti.
Geni Essenziali e Geni Accessori: La Microsintenia
Scendendo a livello dei singoli geni, abbiamo fatto un’analisi di “micro-sintenia”. Abbiamo raggruppato tutti i geni (oltre 830.000!) di 30 (sub)genomi Oryza e dell’outgroup Leersia in circa 77.000 “cluster sintenici”, cioè gruppi di geni omologhi che si trovano in posizioni corrispondenti nei diversi genomi. Questo ci ha permesso di costruire un “pangenoma sintenico”, una sorta di catalogo completo dei geni della famiglia Oryza, distinguendo tra:
- Geni “core”: presenti in tutti (o quasi tutti, “softcore”) i (sub)genomi. Sono solo una piccola parte (circa 6.256 cluster, l’8.1%), ma svolgono funzioni essenziali, quelle che non possono mancare.
- Geni “dispensabili”: presenti solo in alcune specie o gruppi di specie (la stragrande maggioranza, circa 61.130 cluster, il 78.9%). Questi geni sono probabilmente legati ad adattamenti specifici a diversi ambienti o a funzioni meno vitali, e rappresentano la fonte della diversità e della plasticità evolutiva.
- Geni “privati”: trovati in un solo (sub)genoma (pochissimi, 231 cluster, 0.3%).
Questa analisi ci dà un’idea di quali parti del genoma sono fondamentali e quali invece possono essere guadagnate, perse o modificate più facilmente durante l’evoluzione.

Ricostruire l’Albero Genealogico (e Correggere Qualche Errore)
Ovviamente, volevamo capire meglio le parentele tra tutte queste specie. Abbiamo usato due approcci:
- DNA del Cloroplasto: Il cloroplasto (l’organello della fotosintesi) ha un suo piccolo genoma che viene ereditato solo per via materna. Analizzandolo in 26 specie Oryza, abbiamo ricostruito un albero filogenetico che ci dice molto sulle “mamme” ancestrali delle varie linee evolutive.
- Geni Nucleari: Abbiamo usato migliaia di geni “single-copy” (presenti in una sola copia per (sub)genoma) dal nostro pangenoma sintenico per costruire un albero filogenetico molto robusto basato sul DNA del nucleo. Questo albero riflette la storia evolutiva complessiva.
I risultati hanno confermato molte parentele note, ma hanno anche portato a una scoperta importante. Una specie chiamata Oryza schlechteri era classificata con un genoma HHKK. Tuttavia, le nostre analisi (sia filogenetiche che di similarità molecolare, contenuto di TE, ecc.) hanno mostrato in modo inequivocabile che il suo sub-genoma “HH” è in realtà molto più simile al sub-genoma LL di Oryza coarctata (precedentemente riclassificata da HHKK a KKLL) che agli altri HH del complesso ridleyi. Quindi, abbiamo proposto di rinominare O. schlechteri come KKLL, lo stesso tipo di genoma di O. coarctata. È sempre emozionante poter correggere e affinare la classificazione delle specie grazie a dati genomici così dettagliati!
Usando dei punti di calibrazione temporale noti (l’età del genere Oryza, circa 14.5 milioni di anni, e la divergenza tra alcuni gruppi), siamo riusciti anche a datare gli eventi di divergenza tra le specie e a stimare quando sono avvenuti gli incroci che hanno dato origine alle specie tetraploidi (eventi di poliploidizzazione). Ad esempio, le specie BBCC sembrano essersi formate circa 1.73 milioni di anni fa, le CCDD circa 2.54 milioni di anni fa e le HHJJ circa 2.24 milioni di anni fa.
Dopo la Poliploidia: Perdere Geni e Trovare un Equilibrio
Quando due genomi si uniscono per formare un poliploide, la nuova specie si ritrova con un sacco di geni duplicati (le copie provenienti da ciascun genitore, chiamate omeologhe). Col tempo, però, molti di questi geni extra vengono persi, in un processo chiamato frazionamento genico. È come se il genoma cercasse di tornare a uno stato più “snello”.
Ci siamo chiesti: questa perdita di geni avviene allo stesso modo nei due sub-genomi ancestrali? A volte sì, a volte no. Abbiamo visto che nelle specie HHJJ (O. ridleyi, O. longiglumis) e in quelle del complesso officinalis (BBCC e CCDD), c’è un frazionamento sbilanciato: un sub-genoma (HH nel primo caso, CC nel secondo) tende a trattenere più geni rispetto all’altro. Abbiamo anche notato che la velocità di perdita dei geni sembra essere maggiore nelle specie poliploidi più “giovani” (le BBCC) e rallentare in quelle più vecchie (HHJJ e CCDD).
Un altro fenomeno interessante nei poliploidi è la dominanza del sub-genoma: a volte, i geni di un sub-genoma tendono ad essere espressi (cioè “accesi” e utilizzati dalla cellula) a livelli più alti rispetto ai loro omeologhi sull’altro sub-genoma. Questo sub-genoma “dominante” tende spesso a subire meno frazionamento. L’alternativa è l’equivalenza dei sub-genomi, dove non c’è una vera dominanza e l’espressione è più bilanciata.
Abbiamo studiato questo aspetto in Oryza coarctata (KKLL), una specie super interessante perché è alofita, cioè tollera alte concentrazioni di sale. Qui, nonostante avessimo osservato un frazionamento leggermente sbilanciato (il sub-genoma KK perdeva un po’ più di geni del LL), analizzando l’espressione genica in foglie e radici abbiamo scoperto una cosa affascinante: non c’era una vera dominanza! Alcuni geni omeologhi erano più espressi nel sub-genoma KK, altri nel sub-genoma LL, in una sorta di mosaico che suggerisce equivalenza tra i due sub-genomi. Inoltre, abbiamo confermato un’ipotesi generale: i geni che vengono persi più facilmente (frazionati) tendono ad essere quelli che erano già espressi a livelli più bassi, mentre i geni altamente espressi hanno maggiori probabilità di essere mantenuti in doppia copia.

Un Tesoro per il Futuro
Questo enorme set di dati genomici che abbiamo generato e analizzato è molto più di un semplice esercizio accademico. È una risorsa preziosissima per il futuro. Ci permette di:
- Capire più a fondo l’evoluzione dei genomi vegetali, specialmente dopo la poliploidia.
- Scoprire geni utili nei parenti selvatici del riso (per la tolleranza a stress, resistenza a malattie, qualità nutrizionali) da poter introdurre nel riso coltivato tramite incroci o tecniche di editing genetico.
- Esplorare la “neodomesticazione”: provare a domesticare da zero alcune di queste specie selvatiche già adattate a climi difficili.
- Studiare la genetica di popolazione delle specie selvatiche per capire come conservare al meglio la loro diversità genetica, fondamentale per il futuro del nostro pianeta.
Insomma, abbiamo aperto una finestra incredibile sulla storia e sul potenziale del genere Oryza. È come avere una mappa del tesoro molto più dettagliata, che ci guiderà verso soluzioni innovative per l’agricoltura di domani. Il viaggio è appena iniziato!
Fonte: Springer





