Cellule Sotto la Lente: Come l’Ambiente Tissutale Cambia le Regole del Gioco Genetico
Il Grande Puzzle della Diversità Cellulare
Ciao a tutti! Oggi voglio parlarvi di qualcosa che mi affascina profondamente: come fanno le cellule del nostro corpo, pur partendo tutte da un’unica cellula uovo fecondata, a diventare così diverse e specializzate? E soprattutto, come fa l’ambiente in cui vivono, il cosiddetto ambiente tissutale, a influenzare il loro comportamento, in particolare la loro espressione genica? Capire questa diversità è fondamentale per svelare i meccanismi complessi che regolano i sistemi biologici.
Negli ultimi anni, grazie a tecnologie potentissime come il sequenziamento dell’RNA a livello di singola cellula (single-cell RNA-seq), abbiamo iniziato a creare delle vere e proprie mappe, degli “atlanti omici”, che raccolgono dati da tantissimi tessuti e tipi cellulari diversi. Questi atlanti sono miniere d’oro di informazioni, ma c’è un problema che ci ha sempre un po’ frenato: isolare l’effetto *puro* dell’ambiente tissutale è incredibilmente difficile.
La Sfida del Confondimento Statistico
Perché è così complicato? Immaginate di voler capire se vivere in montagna (tessuto A) o al mare (tessuto B) influenzi l’altezza (espressione genica) delle persone (cellule). Il problema è che magari in montagna vivono prevalentemente persone di un certo tipo (tipo cellulare X) e al mare persone di un altro tipo (tipo cellulare Y). Come fate a sapere se la differenza di altezza dipende dal luogo o dal tipo di persona? Questo è quello che chiamiamo confondimento statistico.
Nel corpo umano (o, nel nostro caso di studio, nel topo), la situazione è simile: alcuni tipi di cellule, come quelle del sangue o i fibroblasti, si trovano in molti tessuti, ma tante altre sono specifiche di un solo organo (pensate agli epatociti nel fegato). Inoltre, non sempre riusciamo a raccogliere tutti i tipi cellulari da ogni tessuto. Il risultato è che le combinazioni “tessuto-tipo cellulare” che possiamo osservare sono solo una piccola frazione di tutte quelle teoricamente possibili. Questo “buco” nei dati crea un enorme confondimento tra l’effetto del tessuto e quello del tipo cellulare, rendendo difficile capire cosa influenzi davvero l’espressione di un gene.
Quantificare l’effetto isolato del tessuto è però cruciale. Se un gene si esprime in tutte le cellule di un certo tessuto, ma non altrove, significa che qualcosa in quell’ambiente (magari delle nicchie cellulari particolari o proteine secrete) sta giocando un ruolo chiave. Se invece un gene si esprime in un certo tipo cellulare in tanti tessuti diversi, allora probabilmente è qualcosa di interno alla cellula stessa, come il suo stato epigenetico, a comandare.
COSER: La Nostra Arma Segreta Contro il Confondimento
Di fronte a questa sfida, abbiamo pensato: “E se potessimo trovare dei ‘mini-mondi’ all’interno dei grandi atlanti di dati, dove tutte le combinazioni che ci interessano sono presenti?”. Ed è qui che entra in gioco la nostra nuova creatura: un framework di analisi che abbiamo chiamato COSER (Combinatorial Sub-dataset Extraction for Confounding Reduction).
L’idea alla base di COSER è usare la teoria dei grafi. Immaginate di rappresentare i tessuti e i tipi cellulari come due insiemi di punti, e di tracciare una linea (un “arco”) tra un tessuto e un tipo cellulare solo se quella combinazione esiste nel nostro dataset. COSER va a caccia, all’interno di questo grande grafo, di sottografi speciali chiamati “biclique” (o loro estensioni a più variabili, come sesso, età, ecc., che tecnicamente chiamiamo soluzioni in ipergrafi k-partiti). Una biclique è un gruppo di tessuti e un gruppo di tipi cellulari tale per cui *tutte* le possibili combinazioni tra i membri dei due gruppi esistono nel dataset.
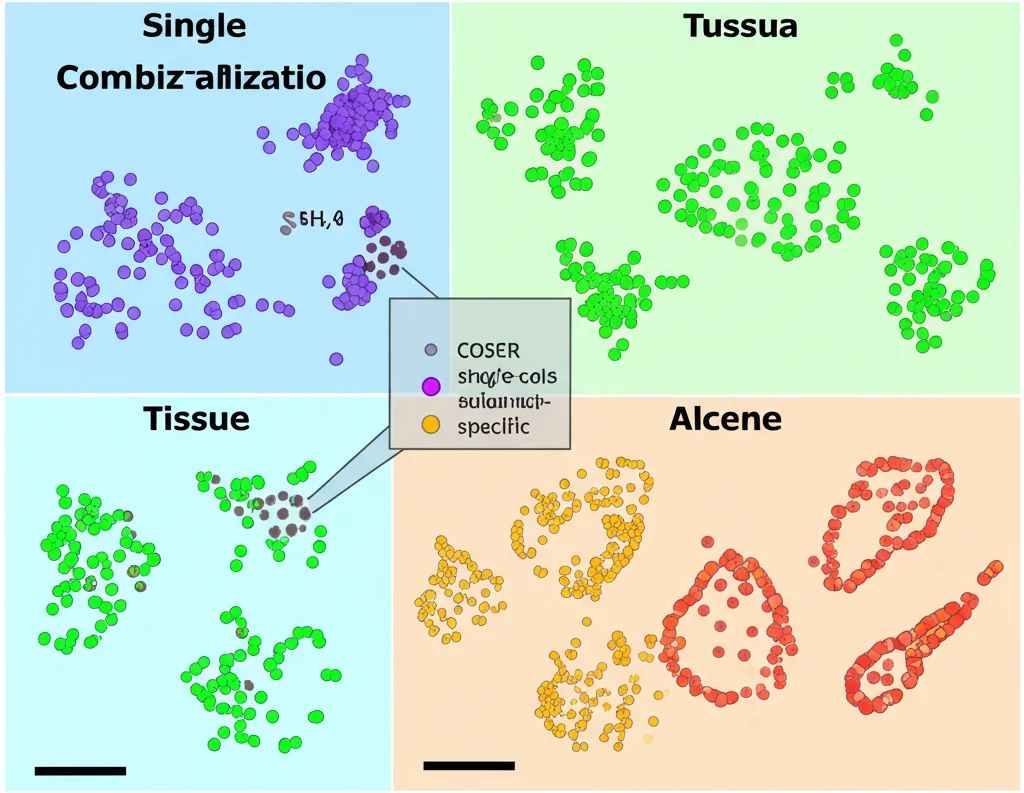
Analizzando separatamente i dati corrispondenti a ciascuna di queste “biclique” (che chiamiamo sub-dataset), possiamo usare modelli statistici (come i modelli lineari generalizzati, GLM) per valutare l’effetto di ogni variabile (tessuto, tipo cellulare, età…) senza il fastidioso confondimento. È come fare tanti piccoli esperimenti controllati all’interno di un grande set di dati osservazionali!
Cosa Abbiamo Scoperto Applicando COSER
Abbiamo messo alla prova COSER su un dataset pubblico fantastico, il Tabula Muris Senis (TMS), un atlante single-cell RNA-seq di topi di diverse età (da 1 a 30 mesi) e che copre ben 23 tessuti. Abbiamo analizzato separatamente i dati ottenuti con due tecniche diverse (FACS e Droplet).
Concentrandoci sui topi di 3 mesi e considerando le combinazioni di individuo, tessuto e tipo cellulare, COSER ha identificato 24 “sub-dataset” ideali nel set di dati FACS e uno nel set Droplet. Analizzando questi sub-dataset, abbiamo avuto la conferma: l’ambiente tissutale ha un effetto isolato e significativo sull’espressione genica, proprio come il tipo cellulare!
Quali geni sono più sensibili all’ambiente? Abbiamo identificato 253 geni nel dataset FACS il cui livello di espressione era significativamente influenzato dal tessuto in almeno metà dei sub-dataset analizzati. E qui viene il bello: facendo un’analisi di arricchimento funzionale (Gene Ontology), abbiamo visto che molti di questi geni sono coinvolti in:
- Risposte immunitarie (in particolare alla risposta all’interferone alfa e beta, alla differenziazione delle cellule dendritiche, alla risposta all’interleuchina-4)
- Funzioni cellulari fondamentali come la traduzione citoplasmatica (la sintesi delle proteine)
Risultati simili sono emersi anche dall’analisi del singolo sub-dataset Droplet (che confrontava muscolo dell’arto e ghiandola mammaria), suggerendo che questi processi sono influenzati dall’ambiente tissutale in tutto il corpo.
Un Focus sui Tessuti Adiposi e l’Invecchiamento
COSER ci ha permesso anche di fare confronti più sottili. Ad esempio, abbiamo potuto confrontare l’effetto ambientale tra diversi tipi di tessuto adiposo (grasso bruno – BAT, grasso gonadico – GAT, grasso mesenterico – MAT, grasso sottocutaneo – SCAT). Integrando i risultati delle analisi sui vari sub-dataset, siamo riusciti a costruire dei grafici (DAG, Directed Acyclic Graphs) che mostrano un ordine gerarchico dell’effetto ambientale su alcuni geni. Per esempio, per il fattore di trascrizione Tbx15 (importante per il grasso bruno), l’effetto ambientale segue l’ordine SCAT > BAT > GAT > MAT. Per Wt1 (espresso nei progenitori del grasso viscerale), l’ordine è GAT > MAT > BAT > SCAT. Queste differenze, note in letteratura, sembrano quindi guidate più dall’ambiente che dal tipo cellulare specifico. Abbiamo trovato risultati interessanti anche per Fosb e Klf4, altri geni legati al grasso.
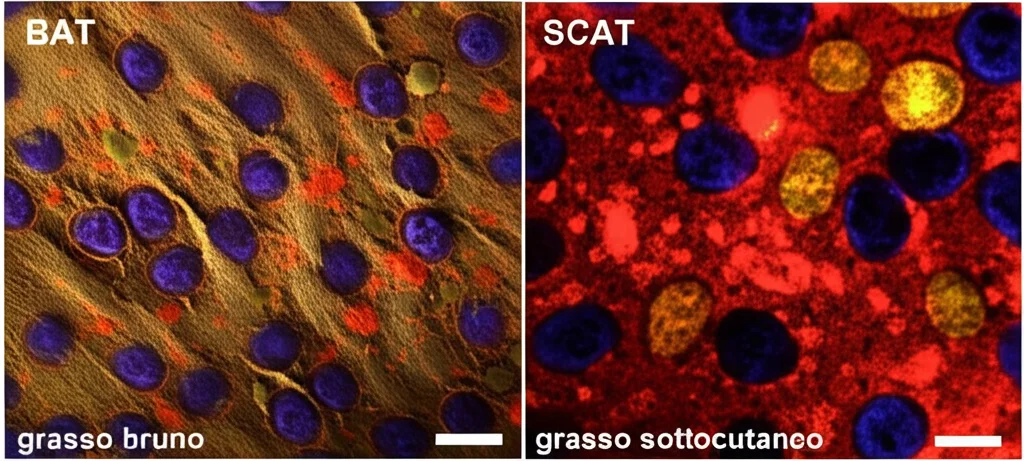
E l’invecchiamento? Sappiamo che l’espressione genica cambia con l’età, ma cambia allo stesso modo in tutti i tessuti? Usando COSER per analizzare combinazioni di sesso, tessuto, tipo cellulare ed età (Giovane vs Vecchio), abbiamo trovato qualcosa di sorprendente confrontando diversi tessuti adiposi nel dataset FACS. Per esempio, tra BAT e SCAT, abbiamo identificato 14 geni che mostravano cambiamenti legati all’età statisticamente significativi ma *in direzioni opposte* nei due tessuti! Molti di questi geni erano legati alla risposta immunitaria e all’infiammazione (difesa dai batteri, metabolismo delle citochine, risposta infiammatoria acuta). Questo suggerisce che l’invecchiamento del sistema immunitario (immunosenescenza) non è un processo uniforme, ma viene modulato in modo diverso a seconda dell’ambiente tissutale. Anche tra MAT e SCAT abbiamo trovato ben 119 geni con effetti opposti legati all’età.
Limiti e Prospettive Future
Ovviamente, come ogni studio, anche il nostro ha dei limiti. Non tutti i tessuti presenti nel dataset TMS sono finiti nelle soluzioni COSER, e alcuni (come il tessuto adiposo) erano sovra-rappresentati. Inoltre, i risultati tra i dataset FACS e Droplet erano diversi, evidenziando come la progettazione dell’esperimento originale influenzi l’applicabilità di COSER. Questo ci dice che COSER potrebbe essere utile anche *prima* di iniziare un esperimento, per pianificarlo al meglio! Infine, ci siamo concentrati molto sui topi di 3 mesi per l’analisi principale. Serviranno più dati, da specie e condizioni diverse, per generalizzare i nostri risultati.
La grande domanda ora è: *come* fa esattamente l’ambiente tissutale a influenzare i geni? Probabilmente attraverso segnali dalle cellule vicine, dalla matrice extracellulare… ma per capirlo serviranno approcci sperimentali mirati.
Quello che abbiamo fatto è stato sviluppare uno strumento, COSER, che ci permette di affrontare il problema del confondimento statistico in modo sistematico quando analizziamo grandi dataset omici con variabili discrete. È un passo avanti importante per studiare gli effetti isolati di fattori come l’ambiente tissutale e capire meglio la straordinaria complessità della vita a livello cellulare. Credo che applicare COSER ad altri tipi di dati omici (epigenomica, proteomica…) potrà svelarci ancora tante sorprese sul paesaggio funzionale delle nostre cellule.
Fonte: Springer