
DNA Aliene nel Batterio Killer? Il Mistero dell’MRSA Cinese tra Cavalli e Topi
Ragazzi, parliamoci chiaro: lo Staphylococcus aureus resistente alla meticillina, meglio noto come MRSA, è un bel grattacapo per la salute globale. È un batterio che non si fa scrupoli, capace di causare infezioni sia negli umani che negli animali, dalle più banali infezioni della pelle a cose serie come polmoniti e sepsi. E sapete qual è una delle sue armi segrete? Una straordinaria capacità di adattarsi e cambiare, una “plasticità genomica” che lo rende un avversario temibile e sempre pronto a evolversi. Capire come si modifica il suo DNA è fondamentale per prevedere le sue mosse future.
Una Scoperta Inquietante: DNA di Cavallo e Topo in Batteri?
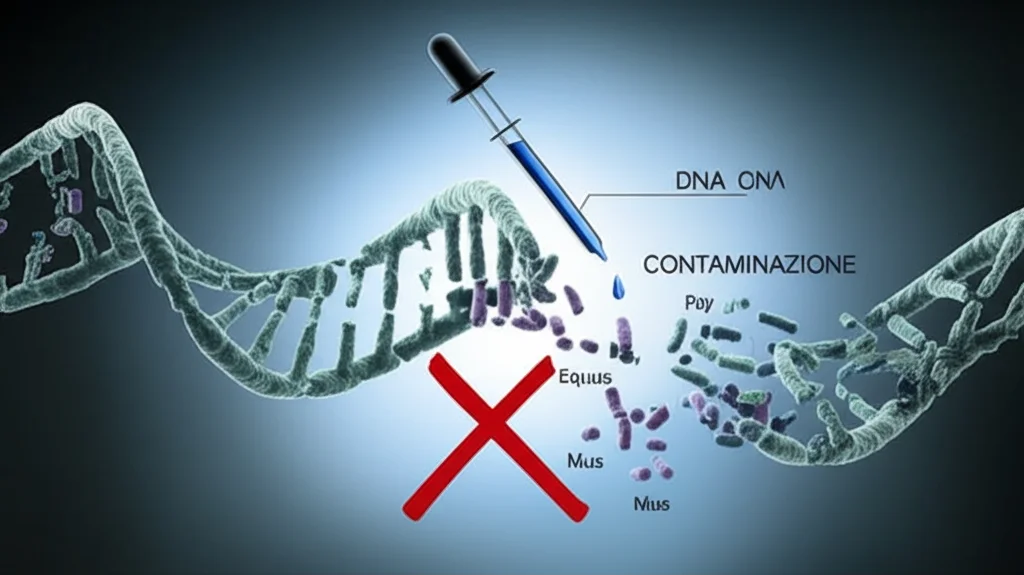
Ma cosa succederebbe se vi dicessi che, sbirciando nel genoma di due ceppi specifici di MRSA isolati a Wuhan, in Cina (chiamiamoli WH3018 e WH9628), abbiamo trovato qualcosa di assolutamente inaspettato? Immaginate la nostra sorpresa nello scoprire delle sequenze di DNA, lunghe da poche centinaia a qualche migliaio di basi, che erano praticamente identiche (fino al 100%!) a pezzi del cromosoma di… Equus caballus (il cavallo) e Mus musculus (il topo)?
Sì, avete capito bene. Nel bel mezzo del genoma di questi batteri, c’erano sequenze che, secondo i database pubblici come GenBank, appartenevano a regioni specifiche del DNA di cavallo (come microsatelliti o regioni del cromosoma Y) o di topo (legate all’RNA ribosomiale 45S-28S o ai loci H19). E la cosa ancora più strana? Questi “frammenti alieni” apparivano più volte, con uno schema ricorrente, in entrambi i ceppi batterici, che tra l’altro sono strettamente imparentati, quasi cloni.
Ricombinazione Interdominio o Errore di Laboratorio?
La prima domanda che ci siamo posti è stata: ma come diavolo è possibile? Una possibilità affascinante, anche se rara, è la ricombinazione interdominio. In pratica, l’idea che il DNA possa saltare da un dominio della vita (come gli eucarioti, cioè animali e piante) a un altro (come i batteri). Sarebbe una scoperta pazzesca, un esempio di trasferimento genico laterale (LGT) su una scala enorme! Sappiamo che circa l’1% dei genomi procariotici potrebbe avere origini eucariotiche, ma trovare pezzi così specifici e recenti di cavallo e topo in un batterio come l’MRSA sarebbe stato rivoluzionario. L’unico caso noto in S. aureus riguarda geni per la resistenza a un antibiotico (mupirocina), ma si pensa che l’evento originale sia antichissimo.
Tuttavia, c’era un’altra possibilità, molto meno esotica ma forse più probabile: la contaminazione dei dati genomici. Il sequenziamento del DNA, specialmente su larga scala (Whole Genome Sequencing – WGS), è un processo complesso. È possibile che, durante la preparazione dei campioni, l’analisi o l’assemblaggio dei dati, minuscoli frammenti di DNA provenienti da altre fonti (magari da studi su cavalli o topi condotti nello stesso laboratorio o con gli stessi reagenti) si siano “infiltrati” nei dati del nostro MRSA?

Gli Indizi Puntano alla Contaminazione
Per cercare di risolvere il mistero, abbiamo fatto diverse analisi.
- Analisi di Ricombinazione: Abbiamo usato software specifici (SplitsTree e RDP4) per cercare le “firme” statistiche della ricombinazione genetica tra il DNA batterico e quello eucariotico. Risultato? Nessun segnale forte e statisticamente robusto che supportasse l’ipotesi di un vero scambio genetico interdominio. Qualche debole suggerimento c’era, ma niente di conclusivo.
- Contenuto G+C: Abbiamo misurato la percentuale delle basi Guanina (G) e Citosina (C) nei frammenti “alieni”. Nel genoma di S. aureus, questo valore è tipicamente basso (circa 32.5%). Nei nostri frammenti sospetti, invece, era significativamente più alto (tra il 47.6% e il 65.0%), più simile a quello che ci si aspetterebbe da DNA eucariotico. Questo rafforzava l’idea che fossero “estranei” al genoma batterico, ma non provava ancora come ci fossero finiti.
- La Natura dei Frammenti: E qui arriva l’indizio forse più pesante. I tipi specifici di DNA di cavallo e topo trovati (microsatelliti, regioni Y, rRNA, H19) sono comunemente usati come marcatori genetici in studi su queste specie. La genotipizzazione, i test di paternità nei cavalli, gli studi su malattie umane usando modelli murini… tutti questi campi di ricerca utilizzano proprio queste sequenze. Il fatto che fossero presenti proprio queste, e da due specie diverse, nei nostri campioni batterici puzzava terribilmente di contaminazione incrociata da campioni o dati di laboratorio. Era uno schema troppo specifico e ricorrente per essere una coincidenza evolutiva.
Un Genoma Batterico Comunque Turbolento
Mentre indagavamo sul mistero del DNA “alieno”, abbiamo notato altre caratteristiche interessanti nei genomi di questi due ceppi MRSA cinesi. Rispetto a un ceppo di riferimento standard (NCTC 8325), i nostri WH3018 e WH9628 avevano:
- Molte più Sequenze di Inserzione (IS): Ben 38 IS per genoma, contro le 16 del riferimento. E non solo di più, ma anche di tipi diversi, alcuni dei quali (IS6, IS21, IS30, IS5/IS1182, IS200/IS605, Tn3) erano presenti solo nei ceppi cinesi. Le IS sono elementi genetici mobili, “saltellano” nel genoma e possono causare mutazioni, riarrangiamenti, e contribuire alla plasticità genomica. È interessante notare che la stragrande maggioranza di queste IS nei ceppi cinesi erano “pseudogeni”, cioè copie non più funzionanti.
- Decadimento Esteso dei Profagi: I profagi sono resti di virus batteriofagi integrati nel genoma batterico. Nel ceppo di riferimento c’erano tre profagi intatti e uno incompleto. Nei ceppi cinesi, invece, trovavamo solo multipli frammenti di profagi, tutti incompleti o “discutibili”. Questo “decadimento” dei profagi potrebbe essere legato a meccanismi di sopravvivenza del batterio.
Queste scoperte suggeriscono che, al di là della probabile contaminazione, i genomi di questi ceppi MRSA sono particolarmente dinamici e “instabili”, forse come risultato della loro storia evolutiva e adattamento.
Contaminazione Anche Altrove?
Un altro dettaglio inquietante: durante le nostre ricerche nei database, abbiamo visto che sequenze simili a quelle trovate nei nostri MRSA (e presunte di cavallo/topo) erano presenti anche in sequenze depositate per altri batteri, come un ceppo di Clostridium culturomicium e uno di Klebsiella pneumoniae. Questo suggerisce che il problema della contaminazione dei dati genomici potrebbe essere più diffuso di quanto pensiamo e merita indagini più approfondite anche su quei genomi.
Lezioni Apprese: L’Importanza di Dati Puliti
Quindi, qual è la morale della favola? Sembra proprio che non abbiamo assistito a un evento fantascientifico di DNA che salta tra cavalli, topi e batteri, ma piuttosto a un caso, seppur complesso, di contaminazione dei dati genomici. Questo studio diventa così un monito importante per tutta la comunità scientifica. Il sequenziamento del genoma intero (WGS) è uno strumento potentissimo per studiare i patogeni, tracciare le epidemie e capire l’evoluzione, ma la qualità dei dati è tutto.
Una contaminazione non rilevata, specialmente se sottile (magari tra ceppi della stessa specie batterica), può portare a conclusioni completamente sbagliate su genotipi, relazioni evolutive, diffusione di resistenze agli antibiotici, mettendo a rischio gli sforzi di sorveglianza e sanità pubblica. C’è un bisogno urgente di linee guida tecniche più avanzate e protocolli standardizzati per garantire l’accuratezza e l’affidabilità dei dati genomici microbici che produciamo e depositiamo nelle banche dati pubbliche.

Per quanto riguarda i nostri ceppi MRSA WH3018 e WH9628, isolati da pazienti con polmonite a Wuhan e appartenenti al tipo ST59 (un clone dominante in Asia), è cruciale rivalutare i loro genomi usando metodologie WGS più robuste per “ripulirli” da queste contaminazioni. Solo così potremo capire veramente la loro biologia, la loro elevata plasticità genomica (indicata dalle tante IS e dai profagi degradati) e il loro potenziale patogeno, contribuendo a controllare meglio la diffusione dell’MRSA in Cina e nel mondo.
Insomma, a volte la spiegazione più semplice (o almeno, la più “terrena”) è quella giusta, ma il percorso per arrivarci può rivelare sfide e complessità inaspettate nel mondo della ricerca genomica!
Fonte: Springer





