Alzheimer: E se la Risposta Fosse in un Computer? La Mia Caccia a Nuove Molecole Miracolose
L’Alzheimer: Una Sfida Complessa che Richiede Nuove Armi
Amici, l’Alzheimer è un osso duro, vero? Una malattia neurodegenerativa progressiva che ci ruba i ricordi, le capacità cognitive e, alla fine, le persone che amiamo. Per anni, noi ricercatori abbiamo cercato la chiave per sconfiggerla, ma la sua natura multifattoriale, cioè causata da tanti piccoli “nemici” che agiscono insieme, la rende particolarmente difficile da affrontare. I farmaci attuali, spesso, prendono di mira un solo colpevole, come l’enzima acetilcolinesterasi (AChE), importante per la trasmissione nervosa, o cercano di contrastare l’accumulo di placche di proteina amiloide-beta (Aβ) nel cervello. Ma diciamocelo chiaramente: non basta. Non abbiamo ancora una cura definitiva, né qualcosa che possa davvero invertire la rotta della malattia.
Ecco perché, nel mio campo, c’è un fermento incredibile verso la ricerca di agenti multifunzionali. Immaginate una sorta di “coltellino svizzero” farmacologico: una singola molecola capace di attaccare l’Alzheimer su più fronti contemporaneamente. E se vi dicessi che la chiave per progettare queste nuove super-molecole potrebbe nascondersi… in un computer? Proprio così! Sto parlando della progettazione in silico, un approccio che sfrutta la potenza di calcolo per disegnare e testare virtualmente nuovi farmaci prima ancora di sintetizzarli in laboratorio. In questo mio ultimo studio, mi sono concentrato su una classe di composti promettenti: i derivati della piridazina.
Perché Proprio le Piridazine? Un Identikit Chimico Promettente
Vi chiederete: “Ma cosa hanno di speciale queste piridazine?”. Beh, la struttura del piridazin-3(2H)-one è considerata una base di partenza fantastica nel design di farmaci, grazie a un’ampia gamma di attività biologiche, tra cui quelle anti-infiammatorie, che non guastano mai quando si parla di neurodegenerazione. Nello specifico, i derivati 2-amminoalchil-6-(2-idrossifenil)piridazin-3(2H)-one che ho studiato combinano diverse caratteristiche farmacoforiche (cioè, le parti della molecola responsabili dell’interazione con i bersagli biologici) davvero interessanti.
- Il gruppo 2-amminoalchile e il nucleo piridazinonico sembrano conferire una buona affinità per l’enzima AChE.
- Il gruppo 2-idrossifenile, invece, può migliorare la solubilità e la stabilità della molecola.
Questa combinazione strutturale non solo ci permette di puntare all’inibizione dell’AChE, ma apre anche la porta alla modulazione di altri bersagli coinvolti nell’Alzheimer, come l’aggregazione della proteina Aβ e lo stress ossidativo. Insomma, un potenziale candidato ideale per la nostra strategia multifunzionale! Già in passato, altri ricercatori hanno usato questa impalcatura strutturale per creare inibitori della colinesterasi, e test in vitro hanno mostrato che molti di questi composti ibridi avevano una forte attività inibitoria contro l’AChE, eccellenti proprietà antiossidanti e una moderata capacità di inibire l’aggregazione di Aβ1-42. Un ottimo punto di partenza, non credete?
Il Mio Arsenale Computazionale: Dalla Teoria alla Progettazione
Per scovare e, soprattutto, progettare nuovi e più potenti derivati piridazinici, ho messo in campo un vero e proprio arsenale di tecniche computazionali. Immaginate di avere a disposizione degli occhiali potentissimi che vi permettono di vedere come una molecola si comporta, come interagisce con le proteine bersaglio e se ha le carte in regola per diventare un farmaco.
Il mio approccio ha incluso:
- Modellazione 2D-QSAR (Quantitative Structure-Activity Relationship): Questa tecnica, un po’ come un detective, cerca di capire quali caratteristiche strutturali di una molecola sono legate alla sua attività biologica. Partendo da un set di 46 molecole piridaziniche già note e dalla loro attività biologica sperimentale (espressa come pIC50), ho costruito un modello matematico.
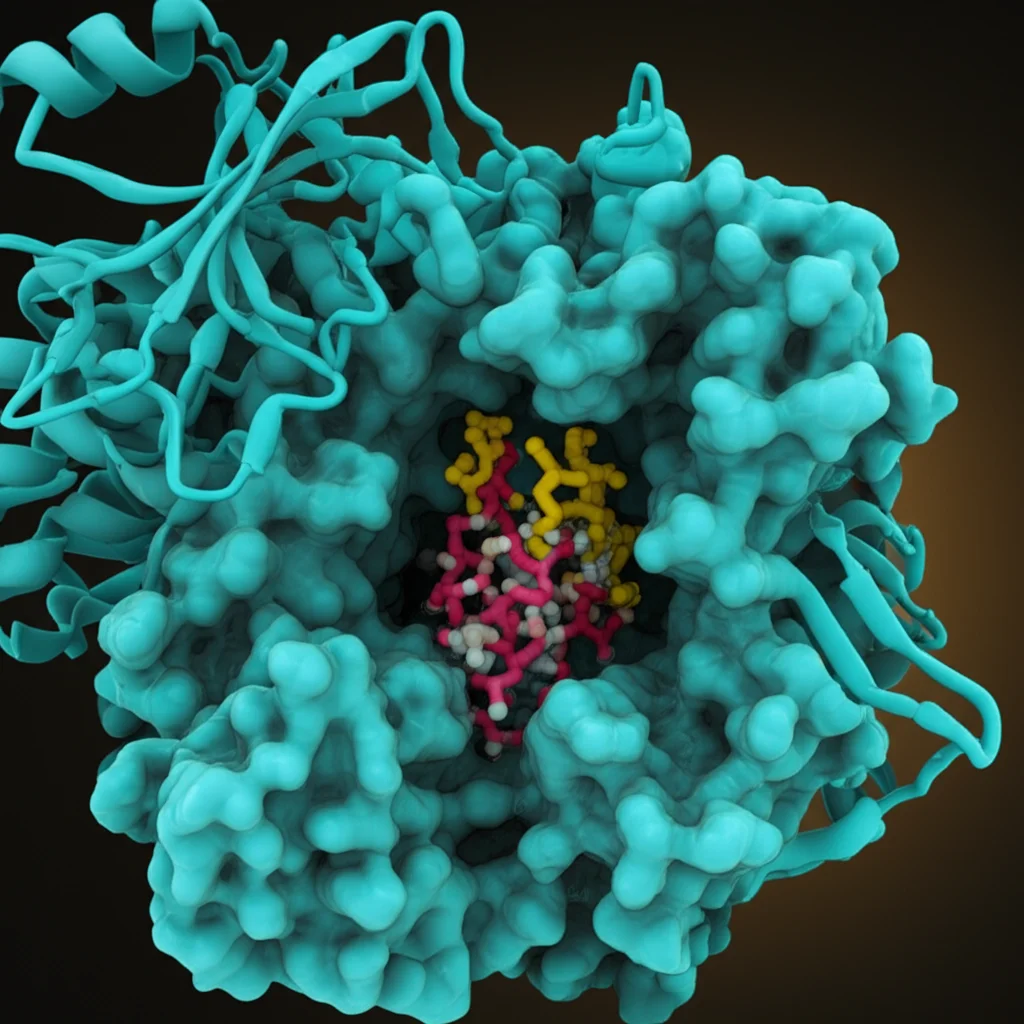
- Molecular Docking: È come cercare la chiave giusta per una serratura. Ho “incastrato” virtualmente le mie molecole (sia quelle note che quelle nuove, da me progettate) nel sito attivo dell’enzima AChE (usando la sua struttura tridimensionale presa da un database, con codice 1EVE.pdb) per vedere quanto bene si legassero e quali interazioni formassero.
- Simulazioni di Dinamica Molecolare (MD): Una volta “dockata” la molecola, non basta. Bisogna vedere se il legame è stabile nel tempo e in condizioni fisiologiche. Le simulazioni MD ci permettono di osservare per un certo periodo (nel mio caso, 100 nanosecondi, che nel mondo molecolare è un tempo significativo!) come il complesso proteina-ligando si muove e si comporta.
- Analisi delle Proprietà ADMET (Assorbimento, Distribuzione, Metabolismo, Escrezione, Tossicità): Un farmaco, per essere efficace, non deve solo legarsi al suo bersaglio, ma deve anche poter essere assorbito dall’organismo, distribuirsi nei tessuti giusti (in questo caso, il cervello!), essere metabolizzato in modo appropriato, essere eliminato e, soprattutto, non essere tossico. Queste analisi predittive sono cruciali per evitare brutte sorprese più avanti.
L’obiettivo era chiaro: usare queste informazioni per progettare nuove piccole molecole derivate dalla piridazina che fossero non solo potenti inibitori, ma anche ottimizzate dal punto di vista farmacocinetico e strutturalmente stabili.

Costruire il Modello QSAR: L’Identikit del Farmaco Ideale
Per il modello 2D-QSAR, ho utilizzato un set di 46 molecole, dividendole in un “training set” (37 molecole per costruire il modello) e un “test set” (9 molecole per validarlo). Ho calcolato ben 31 descrittori molecolari per ciascuna, che spaziano dalla lipofilia agli attributi geometrici e sterici. Alla fine, il modello di regressione lineare multipla (MLR) che ho sviluppato ha identificato cinque descrittori chiave che influenzano significativamente l’attività biologica: S-B (Stretch-Bend), NHBD (Number of Hydrogen Bond Donors), SOVD (Sum of Valence Degrees), TD (Topological Diameter) e TVC (Total Valence Connectivity).
Il modello ha mostrato una buona correlazione (R2 = 0,78) e robustezza statistica. Per capirci, i descrittori S-B, NHBD e SOVD hanno un’influenza positiva sull’attività (più alti sono, meglio è), mentre TVC e TD hanno un’influenza negativa. Ho anche provato con modelli non lineari (MNLR) e reti neurali artificiali (ANN), ottenendo correlazioni ancora migliori (R2 fino a 0,89 con l’ANN), il che mi ha dato grande fiducia nella capacità predittiva del mio approccio. Ovviamente, ho eseguito tutte le validazioni del caso, come la cross-validazione “leave-one-out” (LOO-CV) e la Y-randomization, per assicurarmi che i risultati non fossero dovuti al caso. E, cosa importante, ho verificato il “dominio di applicabilità” del modello con il grafico di Williams, per essere sicuro che le mie previsioni fossero affidabili per le molecole studiate.
Nascita di 13 Nuovi Candidati: Promesse dalla Simulazione
Armato di questo modello QSAR validato, è arrivato il momento più entusiasmante: la progettazione! Basandomi sulle indicazioni fornite dai descrittori molecolari, ho disegnato 13 nuove molecole derivate dalla piridazina (chiamate da Pred1 a Pred13), con l’obiettivo di massimizzare la loro potenziale attività inibitoria contro l’Alzheimer. E le previsioni sono state davvero incoraggianti! Queste nuove arrivate hanno mostrato un’attività inibitoria comparabile o addirittura superiore ai composti più attivi della serie originale.
Ma non mi sono fermato qui. Ho sottoposto questi 13 “eletti” a un’analisi di docking molecolare più approfondita. Ad esempio, i composti C25 e C27 (tra i più attivi della serie originale) e uno dei miei nuovi campioni, Pred12 (che sulla carta sembrava il migliore), hanno mostrato interazioni molto promettenti con residui amminoacidici chiave nel sito attivo dell’AChE, come Trp279, Tyr334 e Phe330. Pred12, in particolare, ha mostrato un’energia di legame bassissima (-7,94 kcal/mol), che suggerisce un legame molto stabile.
Stabilità Dinamica e Proprietà Farmacologiche: Il Test Finale (Virtuale)
Per tre dei ligandi più interessanti (chiamati 61, 726 e 794, simili ai miei Pred), ho eseguito simulazioni di dinamica molecolare per 100 ns. È come guardare un filmato al rallentatore di come la molecola “danza” all’interno della proteina. I risultati? Eccellenti! I complessi proteina-ligando sono rimasti stabili per tutta la durata della simulazione, con fluttuazioni minime (RMSD e RMSF bassi), e hanno mantenuto interazioni cruciali (legami idrogeno, contatti idrofobici) con gli amminoacidi del sito attivo dell’AChE. Questo è un segnale fortissimo che queste molecole non solo si legano bene, ma rimangono anche “agganciate” in modo efficace.
Infine, l’analisi ADMET in silico. Tutti e 13 i miei nuovi composti predetti hanno mostrato un’alta probabilità di essere assorbiti a livello intestinale (circa il 96%!). Hanno rispettato la famosa “regola di Lipinski” (un insieme di criteri per valutare la “drug-likeness”, cioè quanto una molecola assomiglia a un farmaco) e i requisiti di area superficiale polare topologica (TPSA) per i farmaci che agiscono sul sistema nervoso centrale. Inoltre, si prevede che abbiano una moderata solubilità in acqua e una buona capacità di assorbimento gastrointestinale. Dal punto di vista del metabolismo, sembrano essere substrati e inibitori del citocromo CYP3A4 (un enzima importante nel metabolismo dei farmaci) e mostrano valori di clearance (velocità di eliminazione dal corpo) promettenti. E la ciliegina sulla torta: nessuno di loro è risultato tossico nelle valutazioni preliminari!
Conclusioni e Sguardo al Futuro: Una Nuova Speranza?
Cosa ci portiamo a casa da tutto questo lavoro al computer? Beh, direi parecchio! Abbiamo identificato cinque descrittori molecolari chiave che influenzano l’attività biologica dei derivati piridazinici contro l’Alzheimer. E, cosa più importante, abbiamo progettato in silico 13 nuove molecole che non solo promettono un’elevata attività inibitoria, ma mostrano anche caratteristiche farmacocinetiche e di sicurezza favorevoli. Queste molecole hanno il potenziale per agire come agenti multifunzionali, colpendo sia l’AChE che l’aggregazione di Aβ, e quindi potrebbero essere preziose per ritardare la progressione della malattia.
Certo, siamo ancora nel regno delle simulazioni. Il prossimo passo, fondamentale, sarà sintetizzare questi composti in laboratorio e testarli in vitro e poi, speriamo, in vivo. Ma i risultati ottenuti finora sono una solida base di partenza e offrono a noi chimici farmaceutici tantissime idee per sviluppare farmaci ancora più potenti. Nel mio futuro, ho già in programma di sviluppare modelli 3D-QSAR per affinare ulteriormente il design di queste terapie.
La strada per sconfiggere l’Alzheimer è ancora lunga e tortuosa, ma ogni piccolo passo avanti, anche quello compiuto grazie alla potenza dei computer, ci avvicina un po’ di più alla meta. E io, nel mio piccolo, sono entusiasta di contribuire a questa caccia!

Fonte: Springer





