Alzheimer: E se Bastasse una Sola Copia Difettosa di un Gene? La Sorprendente Scoperta Finlandese sul TYROBP
Ciao a tutti! Oggi voglio parlarvi di una scoperta che mi ha davvero affascinato e che apre nuove, intriganti prospettive sulla comprensione dell’Alzheimer. Come sapete, questa malattia neurodegenerativa è un puzzle complesso, e la genetica gioca un ruolo cruciale, anche se spesso misterioso. Siamo sempre alla ricerca di nuovi pezzi per completare il quadro, e a volte le risposte arrivano da direzioni inaspettate.
In questo caso, la chiave arriva da una condizione genetica molto rara, la malattia di Nasu-Hakola (NHD), conosciuta anche come osteodisplasia lipomembranosa policistica con leucoencefalopatia sclerosante (PLOSL). Si tratta di una malattia devastante, causata dalla perdita di funzione di *entrambe* le copie dei geni TYROBP o TREM2. Chi ne soffre sviluppa cisti ossee in giovane età, seguite da una demenza precoce di tipo frontotemporale che porta alla morte verso la mezza età.
Il gene TYROBP codifica per una proteina chiamata DAP12, una sorta di “adattatore” che trasmette segnali all’interno delle cellule immunitarie, in particolare della microglia nel cervello. La microglia sono le cellule immunitarie residenti del nostro sistema nervoso centrale, e DAP12 lavora in coppia con recettori importanti come TREM2, giocando un ruolo chiave nella sorveglianza e nella risposta alle patologie cerebrali, inclusa l’Alzheimer. Sappiamo già che alcune varianti del gene TREM2 aumentano il rischio di Alzheimer e demenza frontotemporale. Ma che dire di TYROBP? Finora, il suo ruolo era rimasto piuttosto oscuro, anche perché le varianti potenzialmente dannose di questo gene sono estremamente rare nella maggior parte delle popolazioni studiate.
La Finlandia: Un Laboratorio Genetico Unico
Ed è qui che entra in gioco la Finlandia. La storia unica di questa popolazione, caratterizzata da isolamento relativo e “colli di bottiglia” genetici seguiti da rapida espansione, ha portato a un background genetico più omogeneo, dove alcune mutazioni “fondatrici”, altrimenti rare, sono presenti con una frequenza maggiore. Tra queste, c’è proprio una specifica delezione di 5.2 kilobasi (kb) nel gene TYROBP, che elimina i primi quattro dei cinque esoni del gene. Questa è la delezione che, in omozigosi (cioè quando ereditata da entrambi i genitori), causa la NHD in Finlandia.
Ci siamo chiesti: cosa succede a chi eredita *una sola* copia di questa delezione? Potrebbe essere un fattore di rischio nascosto per malattie neurodegenerative più comuni, come l’Alzheimer, che si manifestano più tardi nella vita? Uno studio precedente aveva esplorato questa possibilità, ma con un numero limitato di portatori identificati (solo 11!), non aveva trovato associazioni significative. Era chiaro che serviva uno studio su scala molto più ampia.
Abbiamo quindi sfruttato la potenza del progetto FinnGen, una biobanca finlandese che combina dati genomici e sanitari registrati per oltre 520.000 individui (circa il 10% della popolazione!). Rilevare grandi delezioni come questa dai dati di genotipizzazione standard (che si concentrano su variazioni di singole lettere del DNA, gli SNV) non è semplice. Ma siamo stati furbi: analizzando il genoma completo di alcuni pazienti NHD finlandesi, abbiamo identificato due SNV specifici che si trovano quasi sempre insieme alla delezione, agendo come “marcatori proxy”. In pratica, cercando questi SNV nei dati FinnGen, potevamo identificare con buona affidabilità i portatori della delezione TYROBP monoallelica (cioè con una sola copia deleta). Ne abbiamo trovati ben 2.231!
La Grande Rivelazione: Rischio Accresciuto di Alzheimer e Demenza
E qui arriva la scoperta principale. Analizzando i dati sanitari di questi individui e confrontandoli con i non portatori, abbiamo fatto centro! È emerso chiaramente che portare una singola copia della delezione TYROBP finlandese è associato a:
- Un aumento significativo del rischio di sviluppare demenza in generale e Alzheimer in particolare (circa il doppio del rischio!).
- Un’età di esordio più precoce per entrambe le condizioni.
Pensate, l’effetto era statisticamente robusto (p < 5x10⁻⁸, la soglia di significatività genome-wide) e, cosa importante, rimaneva significativo anche tenendo conto del notissimo fattore di rischio genetico per l'Alzheimer, l'allele APOEε4. Questo suggerisce che la delezione TYROBP agisce in modo indipendente, aggiungendo un pezzo in più al rischio complessivo di una persona. È la prima volta che viene dimostrata un’associazione così forte a livello dell’intero genoma tra il locus TYROBP e il rischio di AD.
Oltre il Cervello: Anche le Ossa Raccontano una Storia
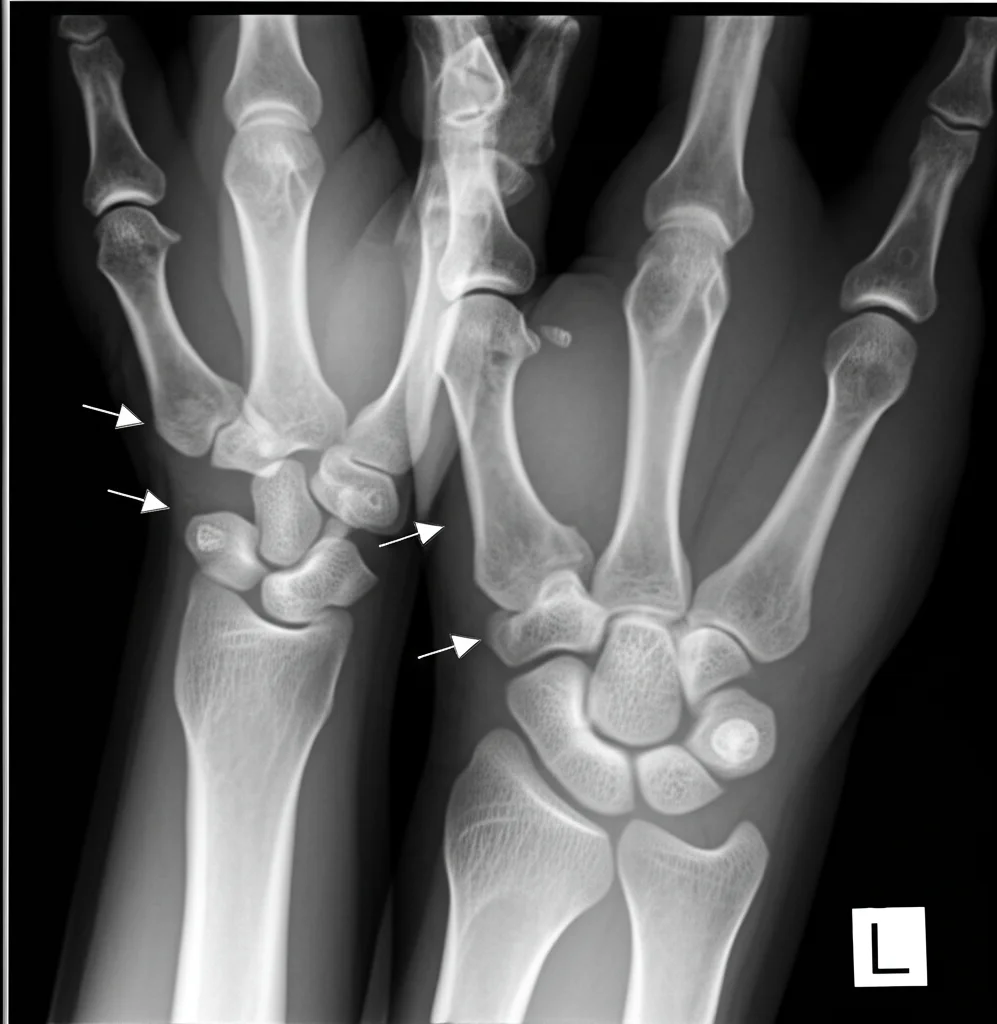
Ma le sorprese non finiscono qui. Ricordate le cisti ossee tipiche della NHD? Abbiamo documentato il caso di una donna di 34 anni, portatrice di una sola copia della delezione TYROBP, che presentava le classiche cisti ossee dolorose ai polsi e alle caviglie, molto simili a quelle viste nei pazienti NHD, ma *senza* alcun sintomo cognitivo o alterazione cerebrale rilevabile alla risonanza magnetica. Il suo fratello, invece, con entrambe le copie delete e diagnosi di NHD, mostrava lesioni ossee molto più estese e gravi, oltre a evidenti segni di atrofia cerebrale e calcificazioni. Questo caso è affascinante perché dimostra che anche una riduzione parziale della funzione di TYROBP/DAP12 può manifestarsi a livello osseo, suggerendo un effetto “dose-dipendente” del gene. Abbiamo anche studiato un’altra portatrice monoallelica, una donna di 75 anni con idrocefalo normoteso e marcatori di patologia Alzheimer nel liquor e nella biopsia cerebrale, ma in questo singolo caso non abbiamo visto differenze nette nella patologia amiloide o nella risposta microgliale rispetto ai non portatori. Serviranno più campioni per capire meglio questo aspetto.
Sbirciare Dentro le Cellule: Cosa Succede a Livello Molecolare?
Per capire *come* la delezione monoallelica eserciti i suoi effetti, siamo passati al laboratorio. Abbiamo prelevato monociti (un tipo di globuli bianchi) dal sangue di portatori monoallelici, pazienti NHD (con delezione biallelica) e controlli sani. Abbiamo poi differenziato questi monociti in cellule “simil-microglia” in coltura (chiamate MDMi).
Prima di tutto, abbiamo confermato quello che ci aspettavamo: le cellule dei portatori monoallelici producevano circa la metà della proteina DAP12 rispetto ai controlli, mentre quelle dei pazienti NHD non ne producevano affatto. Questo conferma che la delezione porta a una ridotta quantità di proteina funzionante.
Poi abbiamo messo alla prova queste cellule MDMi. Le abbiamo lasciate in condizioni normali, oppure le abbiamo stimolate con detriti di mielina (per mimare un danno cerebrale) o con lipopolisaccaride (LPS), una molecola batterica che scatena una forte risposta infiammatoria attivando il recettore TLR4. Utilizzando tecniche avanzate di trascrittomica (analisi dell’RNA) e proteomica (analisi delle proteine), abbiamo osservato cosa cambiava.
Nelle condizioni basali o con mielina, le differenze tra portatori monoallelici e controlli erano minime. Ma con lo stimolo infiammatorio dell’LPS, le cose cambiavano eccome! Le cellule MDMi dei portatori monoallelici mostravano:
- Una risposta infiammatoria potenziata: vie come quella del TNFα via NFKB, la risposta all’interferone gamma e altre vie pro-infiammatorie erano significativamente più attive. Questo è stato confermato misurando le citochine rilasciate: i livelli della citochina pro-infiammatoria IL-1β erano più alti nei portatori monoallelici (e ancora di più nei pazienti NHD) rispetto ai controlli dopo stimolo con LPS.
- Una riduzione della risposta alle proteine mal ripiegate (UPR) e delle vie controllate da MYC. L’UPR è un meccanismo cellulare fondamentale per gestire lo stress causato da proteine danneggiate o accumulate. Una sua disfunzione potrebbe rendere le cellule più vulnerabili. In particolare, abbiamo notato livelli ridotti di PERK (un sensore chiave dell’UPR) e di PSAT1, un enzima coinvolto nel metabolismo e legato, pensate un po’, sia alla funzione immunitaria (regolazione della risposta M2 anti-infiammatoria) che alla differenziazione degli osteoclasti (le cellule che riassorbono l’osso, la cui disfunzione è implicata nelle cisti della NHD). Questa potrebbe essere una connessione molecolare tra i problemi ossei e la risposta immunitaria alterata!
Nei pazienti NHD (delezione biallelica), questi effetti erano ancora più marcati e presenti anche senza stimolo esterno, suggerendo uno stato pro-infiammatorio cronico in queste cellule.

Perché Questa Scoperta è Importante?
Questa ricerca è entusiasmante per diversi motivi. Innanzitutto, identifica la delezione monoallelica di TYROBP come un nuovo fattore di rischio genetico per l’Alzheimer, confermando a livello umano l’importanza della via di segnalazione DAP12 in questa malattia, un ruolo finora suggerito principalmente da studi di network e funzionali.
È interessante notare che studi precedenti su modelli murini di Alzheimer avevano suggerito che ridurre o eliminare Tyrobp/DAP12 potesse essere *protettivo*. I nostri dati sull’uomo dicono esattamente il contrario: una riduzione di DAP12 aumenta il rischio! Questo sottolinea quanto sia cruciale validare le scoperte fatte nei modelli animali con dati umani, data la complessità dell’Alzheimer e le possibili differenze tra specie.
Inoltre, il nostro lavoro mette in luce specifiche vie biologiche alterate dalla riduzione di DAP12 – in particolare l’iper-infiammazione e la disfunzione dell’UPR – che potrebbero rappresentare nuovi bersagli terapeutici. Capire come la mancanza parziale di DAP12 renda le cellule microgliali più “infiammabili” e meno capaci di gestire lo stress proteico potrebbe aprire la strada a strategie per mitigare il rischio associato a questa (e forse altre?) varianti genetiche.
Certo, ci sono delle limitazioni. Lo studio si basa su una specifica delezione trovata nella popolazione finlandese, e replicare questi risultati in altre popolazioni sarà difficile data la rarità di varianti simili altrove. Inoltre, abbiamo usato un modello cellulare (MDMi) che, sebbene utile, non ricapitola perfettamente la complessità della microglia nel cervello umano. Serviranno ulteriori studi, magari usando cellule staminali pluripotenti indotte (iPSC) per creare modelli più sofisticati.
Nonostante ciò, penso che questo studio rappresenti un passo avanti significativo. Ci ricorda che anche geni legati a malattie rare possono insegnarci molto su condizioni comuni come l’Alzheimer, e che esplorare popolazioni geneticamente uniche può portare a scoperte inaspettate. La strada per sconfiggere l’Alzheimer è ancora lunga, ma ogni nuova tessera del puzzle ci avvicina alla meta.
Fonte: Springer