CYP2E1: L’Enzima “Traditore” che Aggrava i Danni al Cuore da Chemioterapia
Ciao a tutti! Oggi voglio parlarvi di una scoperta affascinante, un po’ complessa ma cruciale, che riguarda il nostro cuore e come reagisce a certi trattamenti, come la chemioterapia. Immaginate un farmaco potente come la Doxorubicina (DXR), essenziale per combattere alcuni tumori, ma che purtroppo può avere un lato oscuro: danneggiare il muscolo cardiaco. È un problema serio, noto come cardiotossicità. Per anni, noi ricercatori ci siamo chiesti quali fossero i meccanismi precisi dietro questo danno. Ebbene, sembra che abbiamo trovato uno dei colpevoli nascosti, un enzima chiamato Citocromo P450 2E1, o più semplicemente, CYP2E1.
Chi è questo CYP2E1 e Cosa C’entra col Cuore?
Forse avete già sentito parlare dei citocromi P450. Sono una grande famiglia di enzimi, famosi soprattutto per il loro ruolo nel fegato, dove aiutano a metabolizzare farmaci, tossine e altre molecole. Il CYP2E1 è uno di questi, coinvolto in diverse condizioni patologiche, come i danni al fegato da alcol o la steatoepatite non alcolica. Ma la cosa interessante è che il CYP2E1 non si trova solo nel fegato. È presente anche nel cervello, nei reni, nei polmoni e, appunto, nel cuore.
Negli ultimi anni, abbiamo iniziato a sospettare che questo enzima potesse avere un ruolo anche nelle malattie cardiache. Studi precedenti, inclusi i nostri, avevano suggerito che il CYP2E1 potesse agire come una sorta di “sensore” dello stato di salute del miocardio, il muscolo del cuore. Ma come esattamente influenzasse il danno cardiaco, specialmente quello indotto da farmaci come la DXR, rimaneva un mistero.
La Scoperta Chiave: CYP2E1 nei Mitocondri del Cuore
La vera svolta è arrivata quando abbiamo guardato più da vicino *dove* si trovasse questo enzima all’interno delle cellule cardiache. Tradizionalmente, si pensava fosse principalmente nel reticolo endoplasmatico. Ma analizzando campioni di tessuto cardiaco umano (da pazienti con cardiomiopatia dilatativa o ischemica) e diversi modelli animali di malattie cardiache (indotte da ischemia, ipertensione, farmaci come DXR e Isoprenalina, o anche modelli genetici di cardiomiopatia), abbiamo notato una cosa sorprendente: l’espressione di CYP2E1 aumentava significativamente proprio nei mitocondri delle cellule miocardiche malate.
I mitocondri, ricordiamolo, sono le centrali energetiche delle nostre cellule. Nel cuore, che lavora senza sosta, sono fondamentali. Producono energia (ATP), ma sono anche coinvolti nella gestione dello stress ossidativo, nel controllo della morte cellulare programmata (apoptosi) e nell’omeostasi del calcio. Un loro malfunzionamento è spesso alla base delle malattie cardiache.
Quindi, trovare un aumento specifico di CYP2E1 proprio lì, nei mitocondri del cuore malato, ci ha fatto drizzare le antenne. Poteva essere questa la chiave per capire il suo ruolo nel danno miocardico?

Esperimenti Decisivi: Accendere e Spegnere CYP2E1 nel Cuore
Per verificare la nostra ipotesi, avevamo bisogno di strumenti più precisi. Così, abbiamo fatto qualcosa di piuttosto avanzato: abbiamo creato dei modelli animali (ratti, in questo caso) in cui potevamo controllare specificamente l’espressione di CYP2E1 solo nel cuore. Abbiamo generato ratti in cui il gene Cyp2e1 era “spento” (knockout, KO) nel miocardio e altri in cui era “acceso” più del normale (overexpression, OV).
Una volta ottenuti questi animali “speciali”, li abbiamo sottoposti a stress patologici simili a quelli che causano malattie cardiache nell’uomo:
- Abbiamo somministrato loro la Doxorubicina (DXR) per mimare la cardiotossicità da chemioterapia.
- Abbiamo usato l’Isoprenalina (ISO) per indurre ipertrofia cardiaca patologica (un ingrossamento anomalo del cuore).
I risultati sono stati netti e hanno confermato i nostri sospetti. Nei ratti con CYP2E1 sovraespresso (OV) nel cuore, sia il danno da DXR che l’ipertrofia da ISO erano significativamente peggiori. Questi cuori mostravano pareti più sottili, camere ventricolari più dilatate, funzione cardiaca ridotta, maggiore fibrosi (cicatrici) e disorganizzazione delle fibre muscolari. In pratica, l’eccesso di CYP2E1 accelerava e aggravava la malattia.
Al contrario, nei ratti con CYP2E1 spento (KO) nel cuore, gli effetti dannosi di DXR e ISO erano notevolmente attenuati. Questi cuori erano più protetti, mostravano una migliore funzione cardiaca e minori alterazioni strutturali rispetto ai controlli trattati con gli stessi farmaci. Era la prova che CYP2E1 gioca un ruolo attivo e dannoso in queste condizioni.
Il Meccanismo Svelato: L’Interazione Fatale con OPA1
Ok, CYP2E1 peggiora il danno, ma *come* lo fa, specialmente a livello mitocondriale? Per capirlo, abbiamo usato un mix di tecniche avanzate: analisi trascrittomica (per vedere quali geni venivano influenzati), spettrometria di massa (per identificare le proteine con cui CYP2E1 interagisce), microscopia elettronica (per vedere la forma dei mitocondri) e altre analisi molecolari.
Dall’analisi delle proteine interagenti, è emerso un nome chiave: OPA1 (Optic Atrophy 1). OPA1 è una proteina fondamentale che risiede nella membrana interna dei mitocondri e regola la loro “dinamica”, in particolare la fusione mitocondriale. I mitocondri non sono statici, ma si fondono e si dividono continuamente per mantenere la loro funzionalità. OPA1 esiste in due forme principali: una lunga (L-OPA1), che promuove la fusione, e una corta (S-OPA1), che può derivare dal taglio della forma lunga e avere ruoli diversi, a volte legati alla fissione o alla risposta allo stress. Un equilibrio corretto tra L-OPA1 e S-OPA1 è essenziale per la salute mitocondriale.

Abbiamo scoperto due cose fondamentali:
1. CYP2E1 e OPA1 si trovano nello stesso posto: Utilizzando la microscopia immunoelettronica, abbiamo visualizzato CYP2E1 e OPA1 insieme, proprio sulla membrana mitocondriale interna.
2. CYP2E1 interagisce direttamente con OPA1: Esperimenti di co-immunoprecipitazione hanno confermato che le due proteine si legano fisicamente l’una all’altra.
E l’effetto di questa interazione? Quando CYP2E1 era sovraespresso (come nei cuori malati o nei nostri ratti OV), abbiamo osservato uno squilibrio nel processamento di OPA1: i livelli totali di OPA1 diminuivano e, soprattutto, il rapporto tra la forma lunga (L-OPA1) e la forma corta (S-OPA1) si alterava a favore della forma corta.
Dallo Squilibrio di OPA1 alla Morte Cellulare
Questo squilibrio di OPA1 indotto da CYP2E1 ha conseguenze devastanti. La prevalenza della forma corta e la ridotta fusione portano alla frammentazione mitocondriale. I mitocondri diventano più piccoli, numerosi e disfunzionali. Lo abbiamo visto chiaramente nelle immagini al microscopio elettronico dei cuori dei ratti OV trattati con DXR.
La frammentazione mitocondriale non è solo un problema “estetico”. Mitocondri frammentati e danneggiati sono più inclini a rilasciare nel citoplasma molecole pericolose, come il citocromo c. Il rilascio di citocromo c è un segnale d’allarme che attiva una cascata di eventi molecolari (la via delle caspasi) che porta inesorabilmente alla morte cellulare programmata, l’apoptosi.
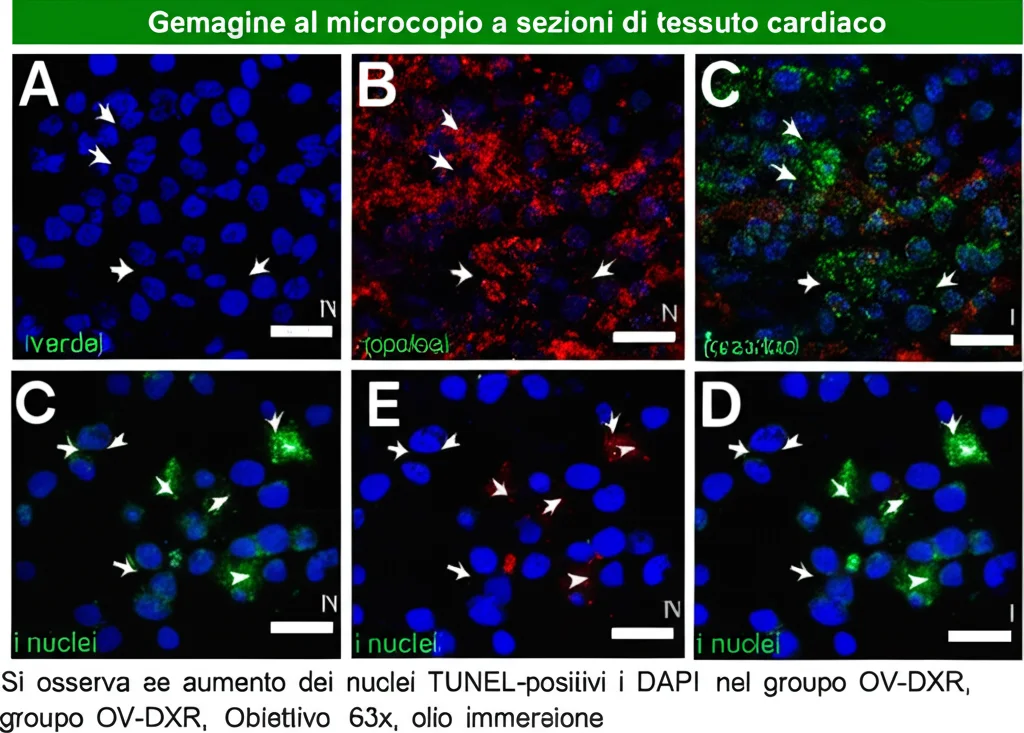
Infatti, nei cuori dei ratti OV-DXR, abbiamo misurato un aumento del rilascio di citocromo c, un’attivazione maggiore della caspasi-3 (l’enzima esecutore dell’apoptosi), un aumento dello stress ossidativo (ROS) e, infine, un numero significativamente più alto di cellule cardiache in apoptosi (misurato con il test TUNEL). Al contrario, nei ratti KO-DXR, tutti questi processi erano ridotti.
Cosa Significa Tutto Questo per Noi?
Questa ricerca ci dice che CYP2E1 non è solo un attore passivo nel cuore, ma un fattore chiave che può peggiorare attivamente il danno miocardico, specialmente in condizioni di stress come la chemioterapia con DXR. Lo fa intrufolandosi nei mitocondri e sabotando l’equilibrio di OPA1, portando a frammentazione mitocondriale, disfunzione e morte delle cellule cardiache.
La buona notizia? Identificare un meccanismo così specifico apre nuove strade terapeutiche. Se riuscissimo a bloccare l’attività di CYP2E1 nel cuore, o magari a impedirne l’interazione con OPA1, potremmo forse proteggere i pazienti dai danni cardiaci indotti da farmaci essenziali come la Doxorubicina. Potrebbe essere una strategia per rendere la chemioterapia più sicura per il cuore.
Certo, la strada dalla ricerca di base alla clinica è lunga, ma aver capito questo intricato meccanismo molecolare è un passo avanti importantissimo. Ci dà un nuovo bersaglio su cui concentrare i nostri sforzi per sviluppare terapie cardioprotettive più efficaci. È la bellezza della ricerca: svelare i segreti della biologia per trovare soluzioni concrete ai problemi di salute. E noi continueremo a scavare!
Fonte: Springer