Caccia Digitale ai Farmaci Antimalarici: La Mia Avventura con le Proteasi Aspartiche!
Ragazzi, parliamoci chiaro: la malaria è ancora un osso duro, una di quelle malattie parassitarie che affliggono l’umanità da tempi immemori e che, purtroppo, continua a essere un problema enorme, specialmente nelle zone tropicali e subtropicali. Milioni di casi ogni anno, centinaia di migliaia di morti, soprattutto tra i bambini piccoli… numeri che fanno rabbrividire. Ecco perché la ricerca di nuovi farmaci antimalarici è una priorità assoluta.
Il Bersaglio: Le Proteasi Aspartiche
Ma come si combatte un nemico così subdolo come il parassita della malaria (il famigerato Plasmodium, nelle sue varie forme come P. falciparum e P. vivax)? Beh, una strategia è colpirlo dove fa più male, mirando a componenti essenziali per la sua sopravvivenza. Uno dei bersagli più promettenti sono le proteasi aspartiche, in particolare una famiglia chiamata plasmepsine. Immaginatele come delle forbici molecolari cruciali per il parassita in diverse fasi del suo ciclo vitale. Se riusciamo a bloccare queste forbici, possiamo fermare il parassita! Studi genetici e farmacologici hanno già confermato che queste proteasi sono talloni d’Achille importanti. Bloccarne alcune, come la plasmepsina IV o IX, ha un impatto devastante sulla sopravvivenza del parassita.
La Sfida della Resistenza e l’Arma Computazionale
Un altro grosso problema è la resistenza ai farmaci. Il parassita è un maestro nel mutare e adattarsi, rendendo inefficaci i farmaci che usiamo oggi. Questo ci spinge a cercare continuamente nuove molecole, nuovi meccanismi d’azione. Ed è qui che entriamo in gioco noi, con i nostri supercomputer e la progettazione di farmaci assistita dal computer (CADD – Computer-Aided Drug Design). Pensateci: invece di testare migliaia di composti a caso in laboratorio (un processo lungo e costosissimo), possiamo usare modelli computazionali per simulare come potenziali farmaci interagiscono con il nostro bersaglio, la proteasi aspartica. È un po’ come usare un simulatore di volo prima di pilotare un aereo vero: ci permette di selezionare i candidati più promettenti in modo molto più rapido ed economico. La CADD accelera enormemente la fase iniziale della scoperta di farmaci, quella che va dall’identificazione del bersaglio alla selezione dei “lead compounds”, le molecole di partenza su cui lavorare.
La Nostra Caccia Virtuale: Docking Molecolare
Nel nostro studio, abbiamo usato proprio queste tecniche computazionali. Il cuore del lavoro è stato il docking molecolare. Immaginate la proteasi come una serratura complessa e i potenziali farmaci come miliardi di chiavi diverse. Il docking ci permette di provare virtualmente quali chiavi (molecole) si adattano meglio alla serratura (il sito attivo della proteasi) e quanto forte è questo “incastro”. Abbiamo preso la struttura tridimensionale della proteasi aspartica del Plasmodium (ottenuta da un database pubblico, il Protein Data Bank, con codice 7TBD) e l’abbiamo preparata per le nostre simulazioni. Poi, abbiamo setacciato due enormi librerie di composti: una di prodotti naturali marini (CMNPD, con circa 32.000 molecole – il mare è una fonte incredibile di nuove sostanze!) e una dal database ZINC (10.000 composti “lead-like”, cioè con caratteristiche promettenti per diventare farmaci).
I Candidati Promettenti
Dopo questa vasta operazione di screening virtuale, sono emerse alcune molecole particolarmente interessanti. Le abbiamo classificate in base alla loro “energia di legame” (binding energy), un punteggio che indica quanto bene si legano alla proteasi (più basso è il punteggio, migliore è l’interazione). Tre composti si sono distinti:
- CMNPD229 (dal database marino) con un punteggio di -8.1 kcal/mol
- ZINC000000018635 con -8.0 kcal/mol
- ZINC000005425464 con -7.8 kcal/mol
Come termine di paragone, abbiamo incluso la clorochina, un farmaco antimalarico noto, che ha ottenuto un punteggio di -6.8 kcal/mol. Vedete? I nostri candidati sembrano legarsi meglio, almeno in silico! Abbiamo visualizzato al computer come queste molecole si incastrano precisamente nel sito attivo della proteasi, formando legami idrogeno e altre interazioni con residui aminoacidici chiave.
Oltre il Docking: Simulazioni e Analisi Approfondite
Ma un buon incastro “statico” non basta. Un farmaco deve funzionare in un ambiente dinamico, quello del nostro corpo. Perciò, abbiamo sottoposto i nostri tre candidati migliori (e la clorochina) a ulteriori test computazionali.
- ADMET: Abbiamo valutato virtualmente le loro proprietà farmacocinetiche (Assorbimento, Distribuzione, Metabolismo, Escrezione) e la potenziale Tossicità. Vogliamo capire se queste molecole hanno la stoffa per diventare veri farmaci: vengono assorbite bene? Raggiungono il bersaglio? Sono sicure? CMNPD229 è risultato particolarmente promettente, mostrando buona assorbibilità, capacità di attraversare la barriera emato-encefalica (importante per alcune forme di malaria) e basso rischio di interferire con il metabolismo di altri farmaci.
- DFT (Density Functional Theory): Siamo scesi a livello quantistico per analizzare la struttura elettronica e la reattività delle molecole. Questo ci dà informazioni sulla loro stabilità e su come potrebbero interagire a livello chimico.
- Simulazioni di Dinamica Molecolare (MDS): Qui la cosa si fa affascinante! Abbiamo creato un “film molecolare” di come la proteasi e il nostro candidato interagiscono nel tempo (per ben 200 nanosecondi, un tempo lunghissimo su scala molecolare!), immersi in un ambiente acquoso simulato a temperatura corporea. Abbiamo analizzato parametri come RMSD e RMSF (che misurano quanto la struttura si discosta e flette nel tempo) e il Raggio di Girazione (RoG, che indica la compattezza). Queste analisi ci hanno mostrato che i complessi formati dai nostri candidati, specialmente CMNPD229, sono molto stabili nel tempo.
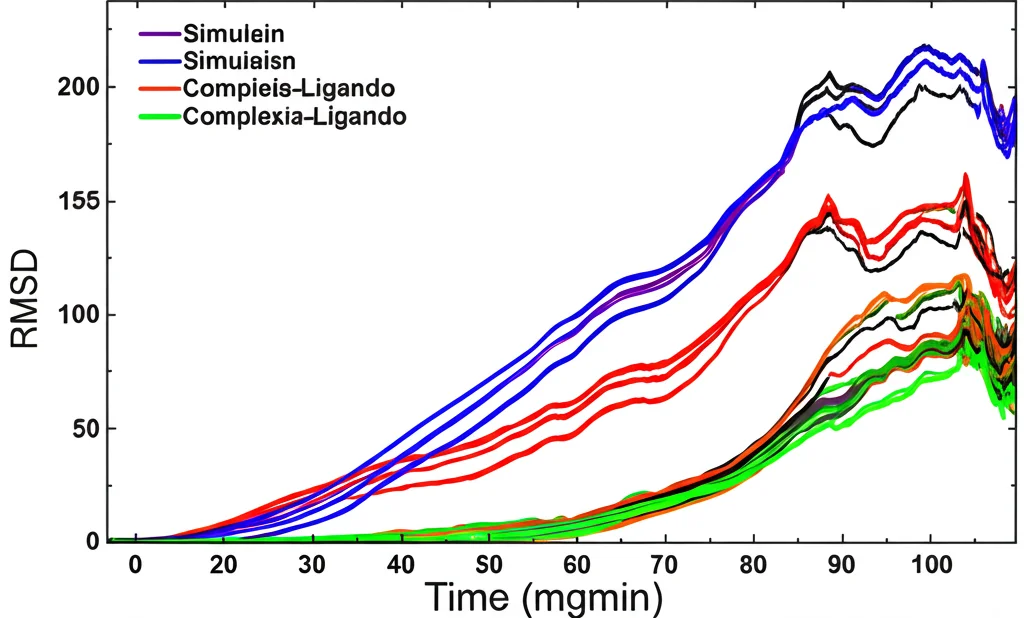
Energia, Entropia e Ponti Salini: La Conferma Finale
Per avere un quadro ancora più completo, abbiamo calcolato l’energia libera di legame usando metodi sofisticati come MM-PBSA/GBSA e WaterSwap. Questi calcoli, che tengono conto di vari fattori energetici (interazioni elettrostatiche, van der Waals, effetti del solvente), hanno confermato che CMNPD229 forma il legame più forte con la proteasi (-120.78 kcal/mol con MM-PBSA/GBSA), seguito da ZINC000000018635 e poi dalla clorochina. Abbiamo anche analizzato l’entropia (il “disordine” del sistema), i ponti salini (forti interazioni elettrostatiche che stabilizzano la struttura) e la struttura secondaria della proteina durante la simulazione. Ancora una volta, CMNPD229 si è distinto, formando un numero maggiore di interazioni uniche (ponti salini specifici), suggerendo una maggiore stabilità e rigidità del complesso. L’analisi PCA (Principal Component Analysis) ci ha aiutato a visualizzare i movimenti collettivi più importanti della proteina, confermando la stabilità conformazionale dei complessi, in particolare quello con CMNPD229.
Conclusioni (Provvisorie) e Prospettive Future
Quindi, cosa ci dice tutta questa fatica computazionale? Ci dice che abbiamo identificato un candidato molto promettente, CMNPD229, un prodotto naturale marino, che sembra essere un potente e stabile inibitore delle proteasi aspartiche del Plasmodium. Ha mostrato ottimi punteggi di docking, proprietà farmacocinetiche favorevoli e una grande stabilità nelle simulazioni dinamiche, legandosi più forte della clorochina (almeno nel nostro modello).
Attenzione però, questo è solo l’inizio! Tutto ciò che abbiamo visto finora è avvenuto dentro un computer. La CADD è uno strumento potentissimo per accelerare la ricerca, ma non sostituisce la validazione sperimentale. Ora la palla passa ai laboratori: bisogna sintetizzare o isolare questi composti e testarli in vitro (su colture cellulari del parassita) e poi, se tutto va bene, in vivo (su modelli animali). Solo così potremo confermare se CMNPD229 e gli altri candidati hanno davvero il potenziale per diventare i nuovi farmaci antimalarici di cui abbiamo disperatamente bisogno. La caccia digitale ha dato i suoi frutti, ora speriamo che la caccia in laboratorio sia altrettanto fortunata!
Fonte: Springer