Autofagia, Stress Mitocondriale e Necroptosi: La Triade Letale Dietro le Malattie Ereditarie della Retina
Ciao a tutti! Oggi voglio parlarvi di un viaggio affascinante nel cuore dei nostri occhi, un viaggio che ci porta a scoprire meccanismi cellulari complessi e, soprattutto, ci dà una nuova speranza per combattere alcune forme di cecità. Parliamo di malattie ereditarie della retina (IRD), un gruppo di patologie che rappresentano una delle principali cause di cecità nel mondo, colpendo sia bambini che adulti in età lavorativa. Pensate, ne soffre circa 1 persona su 1380!
La forma più comune è la retinite pigmentosa (RP), che porta alla perdita progressiva dei fotorecettori, le cellule sensibili alla luce (prima i bastoncelli, poi i coni). Ma ci sono anche altre forme che colpiscono prima i coni o la macula. Il problema grosso? Queste malattie sono incredibilmente eterogenee dal punto di vista genetico: ad oggi conosciamo mutazioni in oltre 280 geni diversi che possono causarle! Questo rende lo sviluppo di terapie, come quelle geniche, una sfida enorme, perché ogni terapia funzionerebbe solo per un piccolo gruppo di pazienti.
Ecco perché la priorità per noi ricercatori è diventata un’altra: trovare dei meccanismi comuni che portano alla morte dei fotorecettori, indipendentemente dal gene mutato. Se capiamo questi processi “a valle”, potremmo sviluppare terapie “agnostiche” rispetto al gene, cioè valide per la maggior parte dei pazienti. Ed è proprio qui che entra in gioco la nostra ricerca.
Indagando cellula per cellula: la potenza della trascrittomica
Per capire cosa succede davvero dentro una retina che sta degenerando, abbiamo usato una tecnica potentissima chiamata trascrittomica a singola cellula (scRNAseq). Immaginate di poter “ascoltare” cosa sta facendo ogni singola cellula della retina, quali geni sta accendendo o spegnendo. L’abbiamo fatto su modelli murini (topi) che mimano la malattia umana, in particolare due modelli con mutazioni nel gene Rpgr, spesso coinvolto nella RP umana. Questi modelli, chiamati RpgrEx3d8 (più rapido) e RpgrORFd5 (più lento), mostrano stress retinico e degenerazione proprio come avviene nell’uomo.
Analizzando migliaia di cellule a 18 mesi di età, abbiamo identificato tutti i principali tipi cellulari della retina. Ma la cosa più interessante è che abbiamo trovato diversi “sotto-gruppi” di fotorecettori (i bastoncelli, in questo caso). Mettendoli in ordine con un’analisi chiamata “pseudotime”, abbiamo visto che questi sotto-gruppi rappresentano una sorta di traiettoria di degenerazione: le cellule passano da uno stato più sano a uno sempre più compromesso, spegnendo i geni fondamentali per la fototrasduzione (il processo di conversione della luce in segnale nervoso).
Analizzando i geni che invece si accendevano lungo questa traiettoria, abbiamo notato un aumento dell’attività legata all’infiammazione (via TNF-α/NF-κB), alla produzione di lisosomi (gli “inceneritori” della cellula) e, cosa molto interessante, alla via di segnalazione PI3K/AKT. Questa via è nota per regolare la sopravvivenza cellulare ma anche l’autofagia, il processo di “pulizia” interna della cellula. Nei fotorecettori mutanti, l’attivazione di questa via era ancora più marcata rispetto ai controlli sani. Abbiamo confermato un aumento della forma attiva di AKT (pAKT) e una diminuzione di alcuni suoi regolatori (PTEN, mTOR) nei topi mutanti, suggerendo che questa via fosse effettivamente iperattiva.
Autofagia in tilt: quando la pulizia non funziona più
L’autofagia è fondamentale: permette alla cellula di eliminare componenti danneggiati o inutili, riciclandone i materiali. È un processo delicatissimo. I nostri dati di scRNAseq mostravano già cambiamenti nei geni legati all’autofagia nei fotorecettori malati. Per vederci più chiaro, siamo passati al microscopio elettronico a trasmissione (TEM), che ci permette di vedere l’ultrastruttura delle cellule.
Nel modello RpgrEx3d8, già a 6 e 12 mesi, abbiamo osservato un accumulo significativo di grosse strutture simili a vescicole nei segmenti interni dei fotorecettori (la parte metabolica della cellula). Sembravano proprio autofagosomi o autolisosomi accumulati, un chiaro segno che l’autofagia non stava funzionando a dovere, come se il sistema di smaltimento fosse bloccato. Questo era confermato dall’aumento di una proteina marcatore dell’autofagia, la p62 (o SQSTM1), e della proteina lisosomiale LAMP1, sempre nei segmenti interni a 12 mesi. È interessante notare che a 18 mesi, quando ormai molti fotorecettori sono morti, questo accumulo sembrava ridursi. Abbiamo anche visto che la p62 era più fosforilata (p-p62 S403), una modifica che di solito ne attiva la funzione autofagica. Quindi, sembrava esserci un tentativo di aumentare l’autofagia, che però finiva per “ingolfarsi”.
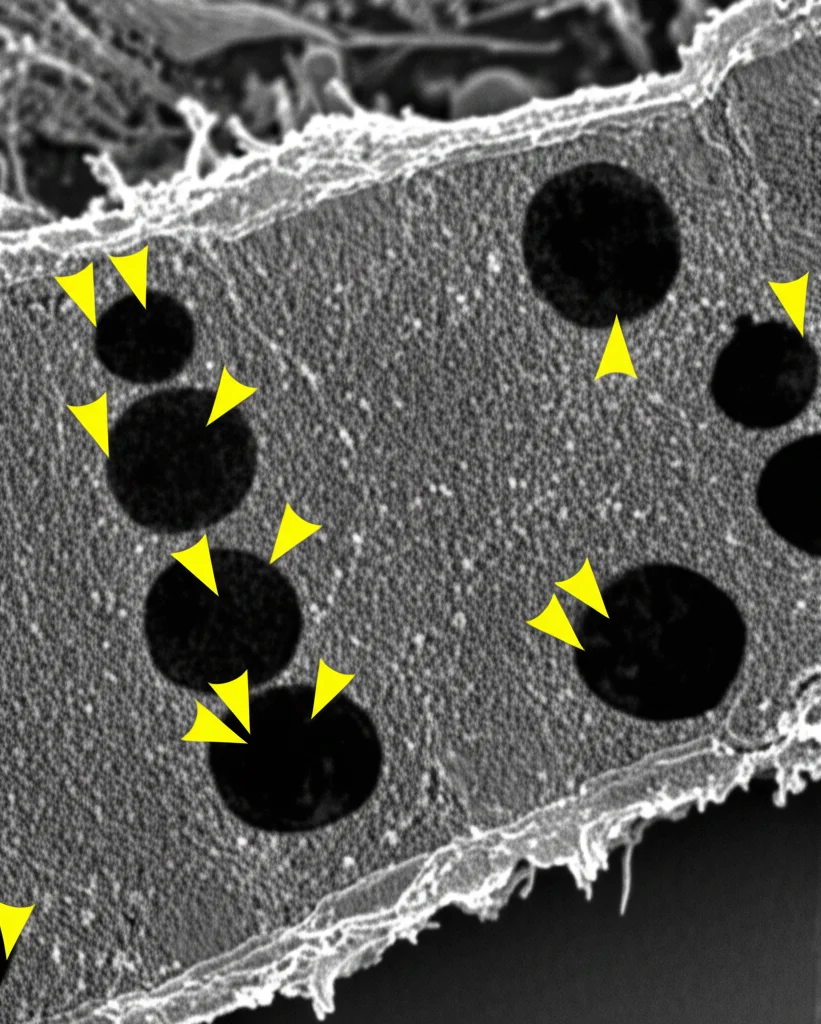
Stress alle stelle: i mitocondri soffrono
Mentre l’autofagia sembrava in difficoltà nelle fasi iniziali e intermedie, cosa succedeva più tardi? Tornando al microscopio elettronico, a 12 mesi abbiamo iniziato a vedere anche problemi ai mitocondri, le nostre centrali energetiche: alcuni apparivano gonfi o con membrane danneggiate. A 18 mesi, questo quadro era molto peggiorato, con un severo gonfiore mitocondriale, chiaro segno di stress mitocondriale.
Anche i dati di scRNAseq supportavano questa osservazione: i geni legati alla funzione mitocondriale e alla loro produzione (biogenesi mitocondriale) erano sovraregolati nei fotorecettori mutanti, forse un tentativo disperato di compensare il danno. Abbiamo confermato l’aumento di alcune proteine della catena di trasporto degli elettroni (CYTC, mtCO1), quelle che producono energia. Inoltre, abbiamo visto un aumento di una proteina chiamata VDAC1 sulla membrana esterna dei mitocondri. VDAC1 è un canale del calcio coinvolto nella risposta allo stress mitocondriale e può promuovere il rilascio di citocromo C, un segnale di morte cellulare.
Per avere una prova diretta della disfunzione, abbiamo misurato il consumo di ossigeno (OCR) in piccoli pezzi di retina prelevati dai topi RpgrEx3d8 a 14 mesi. Il risultato? Un consumo di ossigeno basale significativamente ridotto rispetto ai controlli, confermando che la respirazione mitocondriale era compromessa. I mitocondri dei fotorecettori mutanti erano decisamente sotto stress e non funzionavano più come avrebbero dovuto.

Il colpo di grazia: entra in scena la necroptosi
A questo punto, la domanda era: come muoiono esattamente questi fotorecettori stressati e con l’autofagia in tilt? La morte cellulare programmata più conosciuta è l’apoptosi, una sorta di “suicidio pulito”. Abbiamo cercato i marcatori dell’apoptosi, come la caspasi-3 attivata (cl-casp3). Ne abbiamo trovati alcuni nei mutanti a 18 mesi, ma erano davvero pochi, insufficienti a spiegare la massiccia perdita di cellule. Inoltre, i nostri dati scRNAseq non mostravano una chiara firma trascrizionale apoptotica. Anzi, abbiamo notato una riduzione dell’attività della caspasi-8, un enzima che non solo attiva l’apoptosi ma che, quando è poco attivo, può favorire un’altra forma di morte cellulare: la necroptosi.
La necroptosi è diversa dall’apoptosi. È una morte cellulare regolata, sì, ma molto più “rumorosa” e infiammatoria, caratterizzata dalla rottura della membrana cellulare. E indovinate un po’? L’analisi dei geni differenzialmente espressi nei nostri fotorecettori mutanti puntava proprio verso la necroptosi! Questa via può essere innescata anche dallo stress mitocondriale.
Per confermarlo, abbiamo cercato il marcatore chiave della necroptosi: la proteina MLKL fosforilata (pMLKL). Quando la necroptosi è attiva, pMLKL si accumula sulla membrana cellulare, formando dei pori che la fanno scoppiare. Ebbene, nei nostri modelli Rpgr, abbiamo trovato un numero crescente e significativo di fotorecettori positivi per pMLKL a 6, 12 e 18 mesi, molti di più di quelli positivi per la caspasi-3! Questo aumento seguiva perfettamente la progressione della degenerazione. Sembrava proprio che la necroptosi fosse il meccanismo principale di morte dei fotorecettori in questi modelli.

La prova del nove: la stessa storia in un altro modello di RP
Ok, avevamo trovato questa sequenza di eventi – problemi di autofagia, stress mitocondriale, morte per necroptosi – nei modelli Rpgr. Ma era una cosa specifica di quel gene o un meccanismo più generale? Per scoprirlo, abbiamo esaminato un altro modello murino di RP, completamente diverso: il topo Pde6brd2. Qui la mutazione è nel gene Pde6b, che codifica per una proteina essenziale nella cascata della fototrasduzione, non nella struttura o nel traffico come RPGR. Inoltre, la degenerazione in questo modello è molto più rapida, completandosi entro le prime 4 settimane di vita (P28).
Ebbene, i risultati sono stati sorprendenti e incredibilmente incoraggianti: abbiamo ritrovato esattamente la stessa sequenza di eventi!
- Aumento della necroptosi (pMLKL positiva) che progrediva con la degenerazione.
- Attivazione precoce della via PI3K/AKT (aumento di pAKT).
- Segni di autofagia difettosa (accumulo di autofagosomi al TEM, aumento di p62 e p-p62).
- Evidenza di stress mitocondriale (alterazioni morfologiche al TEM, aumento di VDAC1 e delle stesse proteine della catena respiratoria viste nel modello Rpgr).
Questa conferma in un modello geneticamente e funzionalmente distinto è stata fondamentale. Suggerisce fortemente che l’interruzione dell’autofagia, lo stress mitocondriale e la successiva morte per necroptosi siano davvero meccanismi comuni nella patogenesi della retinite pigmentosa, indipendentemente dalla mutazione genetica scatenante.
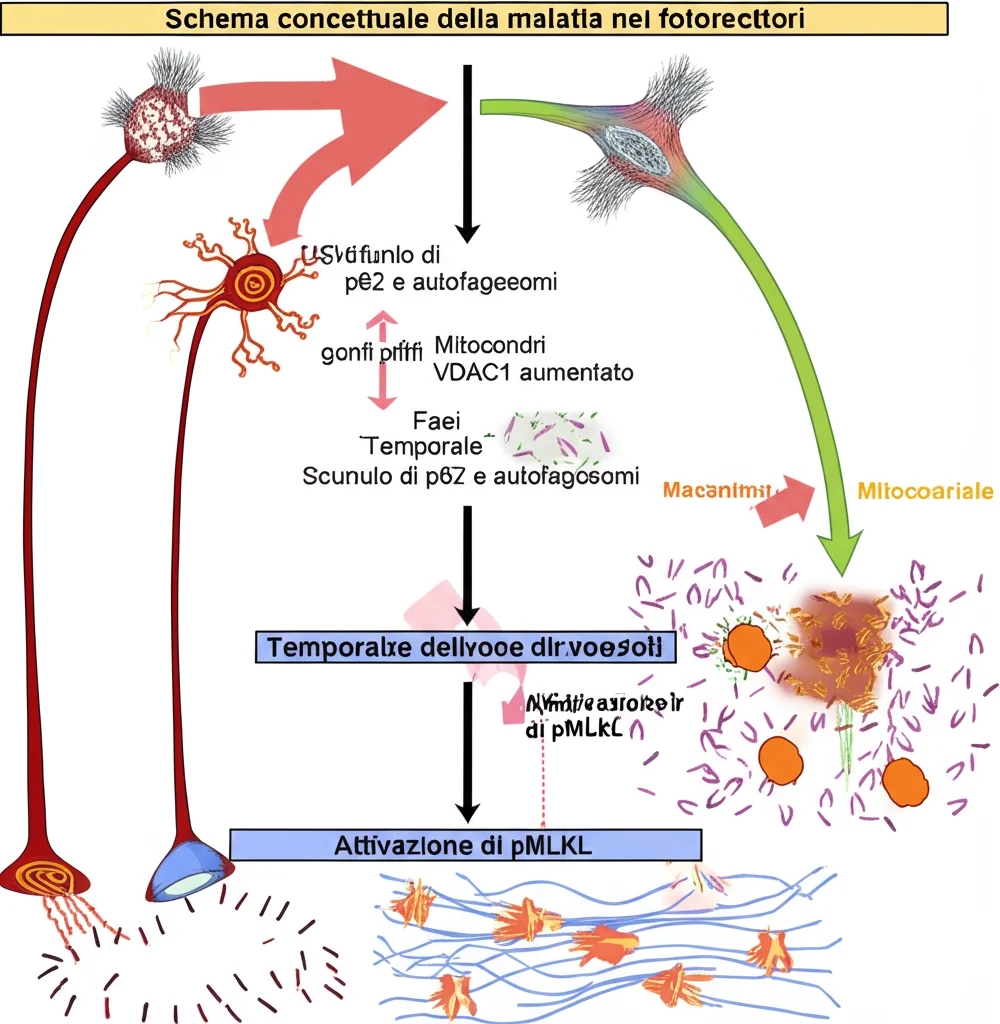
Cosa significa tutto questo per il futuro?
Capire i processi che portano alla morte dei fotorecettori è la chiave per sviluppare nuove terapie. La nostra scoperta che autofagia difettosa, stress mitocondriale e necroptosi sono attori principali in diversi modelli di RP apre scenari terapeutici davvero promettenti. Perché? Perché potremmo pensare a farmaci che agiscano su queste vie comuni, offrendo un trattamento potenziale a molti pazienti, a prescindere dal loro specifico difetto genetico.
Ad esempio, si potrebbe provare a:
- Modulare l’autofagia: anche se è complesso, capire se bisogna riattivarla o inibirla in modo controllato potrebbe essere utile.
- Proteggere i mitocondri: farmaci che riducono lo stress ossidativo o migliorano la funzione mitocondriale potrebbero rallentare la degenerazione.
- Inibire la necroptosi: bloccare questa via di morte infiammatoria sembra un bersaglio molto attraente, e farmaci inibitori della necroptosi sono già in studio per altre malattie.
Certo, la strada è ancora lunga. Bisogna capire meglio l’esatta interazione tra queste tre vie (autofagia/mitofagia, stress mitocondriale, necroptosi) e come si influenzano a vicenda. Ma aver identificato questo percorso comune rappresenta un passo avanti significativo. Ci dà bersagli concreti su cui lavorare per sviluppare terapie innovative e, speriamo, efficaci per preservare la vista in chi soffre di queste devastanti malattie ereditarie della retina. Una speranza che, oggi, sembra un po’ più luminosa.
Fonte: Springer