Il Ballo Segreto dei Recettori: Svelato l’Accoppiamento dei Domini nell’Attivazione dei mGluR
Ciao a tutti! Oggi voglio portarvi con me in un viaggio affascinante nel cuore delle nostre cellule nervose, per scoprire come comunicano tra loro. Avete mai sentito parlare dei recettori metabotropici del glutammato, o mGluR? Sono proteine incredibilmente importanti che si trovano sulla superficie dei neuroni e giocano un ruolo chiave nella trasmissione dei segnali e nella plasticità sinaptica – in pratica, sono fondamentali per come impariamo e ricordiamo!
Questi recettori non lavorano da soli, ma formano coppie chiamate dimeri (omomeri, se i partner sono identici, o eteromeri, se sono diversi). La cosa pazzesca è che, a seconda di come si accoppiano, rispondono in modo diverso al glutammato, il loro “messaggero” chimico principale. Ma come fanno esattamente ad attivarsi? Come fa il legame del glutammato a innescare una risposta all’interno della cellula? È proprio quello che abbiamo cercato di capire.
La Struttura Complessa degli mGluR
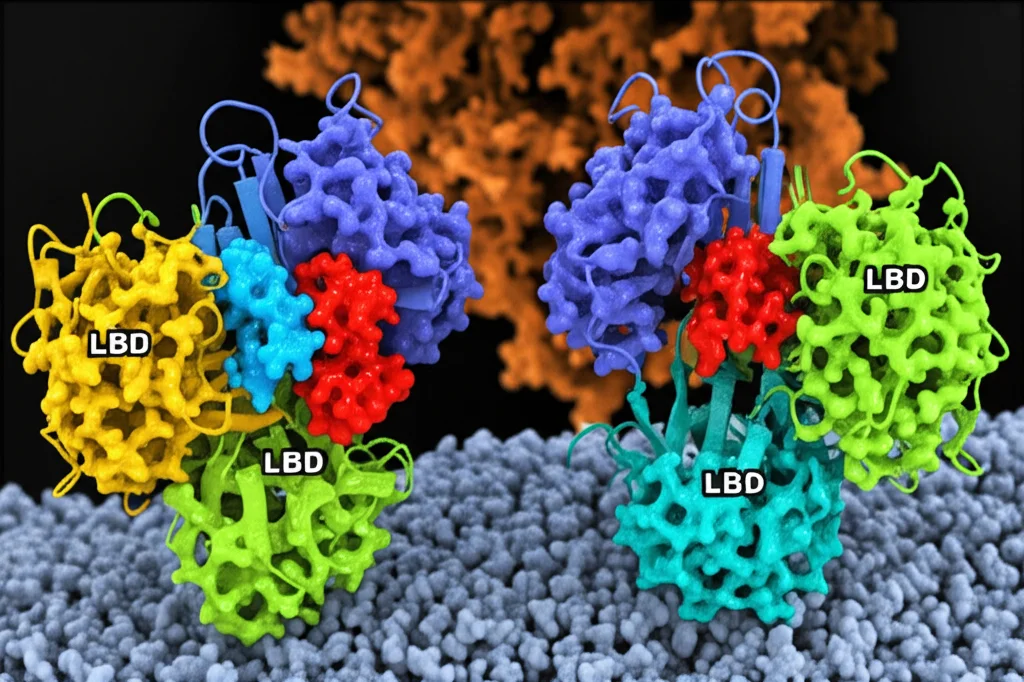
Immaginate questi recettori come delle complesse macchine molecolari. Ogni “partner” (subunità) ha tre parti principali:
- Una grande porzione esterna alla cellula, chiamata dominio legante il ligando (LBD), che assomiglia un po’ a una “trappola per Venere” (Venus Flytrap) e che afferra il glutammato.
- Una parte che attraversa la membrana cellulare sette volte, il dominio transmembrana (TMD).
- Un piccolo dominio ricco di cisteina (CRD) che fa da ponte tra LBD e TMD.
Quando il glutammato si lega, l’LBD si chiude come una conchiglia e i due LBD nel dimero “ruotano” l’uno rispetto all’altro. Questo movimento si trasmette attraverso i CRD fino al TMD, che cambia forma all’interno della cellula e attiva altre proteine, le proteine G, dando il via alla segnalazione. Sembra semplice, no? Beh, la realtà è molto più intricata e dinamica!
Spiare i Recettori in Azione: la Magia dell’smFRET
Per vedere questi movimenti nel dettaglio, abbiamo usato una tecnica super potente chiamata spettroscopia a singola molecola FRET (smFRET). In pratica, attacchiamo delle minuscole “luci” fluorescenti (un donatore e un accettore) a punti specifici del recettore. Quando questi punti si avvicinano, l’energia passa dal donatore all’accettore, che si illumina di più (segnale FRET alto). Misurando questo segnale nel tempo per singole molecole, possiamo letteralmente “vedere” i domini muoversi!
Abbiamo messo le nostre “luci” per monitorare due movimenti chiave:
- La chiusura della “conchiglia” LBD su una singola subunità.
- La “torsione” (twisting) tra i due LBD nel dimero.
Cosa abbiamo scoperto? Che queste due mosse, chiusura e torsione, non sono perfettamente sincronizzate! Anche quando c’è tantissimo glutammato e le conchiglie LBD sono quasi sempre chiuse, il dimero LBD non rimane fisso nella conformazione “attiva” (ruotata), ma continua a passare da uno stato all’altro. È come se la chiusura fosse necessaria ma non sufficiente per bloccare il recettore in posizione attiva. Questo suggerisce che l’attivazione è un processo a più passi, con un accoppiamento “lasco” tra i movimenti dei domini.
Ancora più interessante: abbiamo visto che l’aggiunta di molecole chiamate modulatori allosterici positivi (PAM), che si legano al TMD, o della proteina G stessa, spingeva il dimero LBD a rimanere più a lungo nella conformazione attiva (ruotata). Questo ci dice che c’è una comunicazione “verticale” lungo il recettore: quello che succede nel TMD influenza l’LBD, e viceversa!
Zoom Molecolare: Simulazioni al Computer e Mutazioni Mirate
Per capire quali fossero le forze molecolari dietro questa danza, siamo passati alle simulazioni di dinamica molecolare (MD). Abbiamo preso la struttura 3D del dimero LBD attivo e l’abbiamo messa in un ambiente virtuale che simulava le condizioni cellulari, lasciandola “muoversi” per tempi lunghissimi (microsecondi!).
Ebbene, le simulazioni ci hanno mostrato una cosa sorprendente: anche partendo dalla forma attiva, il dimero LBD a volte “rilassava” la sua torsione, pur mantenendo le conchiglie chiuse! Abbiamo chiamato questo stato intermedio “Rilassato-Chiuso/Chiuso” (R-C/C). Analizzando le interazioni tra gli amminoacidi durante queste transizioni, abbiamo identificato una rete di contatti, in particolare un ponte salino tra i residui Arginina 177 (R177) e Aspartato 95 (D95) di subunità opposte, che sembrava cruciale per mantenere la torsione attiva.
Siamo tornati agli esperimenti smFRET e abbiamo creato versioni mutate del recettore mGluR2, sostituendo R177 o D95 con un amminoacido neutro (Alanina). E indovinate un po’? Queste mutazioni riducevano drasticamente la capacità del recettore di raggiungere e mantenere la conformazione LBD attiva (ruotata), anche con un agonista molto potente! Questo conferma che quella rete di interazioni è davvero una sorta di “chiavistello” molecolare che regola l’efficacia dell’attivazione.
Dal LBD al TMD: il Ruolo dei Linker CRD e l’HDX-MS
Ma come si propaga il segnale dall’LBD al TMD? Abbiamo messo le nostre “luci” FRET anche sui domini CRD, i linker che collegano LBD e TMD. Abbiamo visto che, in risposta al glutammato, anche i CRD si avvicinano tra loro, ma questo movimento è ancora più “pigro” di quello dell’LBD! Sembra che l’avvicinamento dei CRD avvenga dopo la torsione dell’LBD, suggerendo un altro passo con accoppiamento lasco.
Solo quando abbiamo aggiunto un PAM (BINA) o la proteina G, i CRD hanno iniziato a popolare stati ancora più vicini tra loro, uno dei quali corrisponde proprio alla distanza vista nelle strutture del recettore legato alla proteina G. Tuttavia, anche in queste condizioni, il recettore continuava a fluttuare tra diversi stati di vicinanza dei CRD. Questo significa che la conformazione pienamente pronta per segnalare (quella che lega la proteina G) è occupata solo per una piccola frazione del tempo, anche quando il recettore è stimolato. C’è quindi un ampio “margine di manovra” per la modulazione da parte di PAM e altre molecole.

Per avere un quadro ancora più completo della flessibilità del recettore, abbiamo usato l’HDX-MS (scambio idrogeno-deuterio monitorato da spettrometria di massa). Questa tecnica misura quanto facilmente gli atomi di idrogeno lungo la spina dorsale della proteina possono scambiarsi con il deuterio (un isotopo pesante dell’idrogeno) presente nell’acqua circostante. Le regioni più flessibili o esposte scambiano più velocemente.
Abbiamo confermato che il glutammato “protegge” dallo scambio le regioni del sito di legame e alcune eliche (C, D, F) nell’interfaccia di dimerizzazione dell’LBD, rendendole meno flessibili. Confrontando il recettore completo con il solo LBD, abbiamo visto che la presenza del TMD e del CRD stabilizza alcune parti dell’interfaccia LBD anche senza glutammato. E l’aggiunta del PAM BINA? Rendeva meno flessibile soprattutto l’elica F nell’LBD, un’elica che fa da ponte fisico tra il sito di legame del glutammato e il CRD. Questo suggerisce un meccanismo attraverso cui i modulatori nel TMD possono “risalire” e influenzare l’LBD.
Compagni Diversi, Danze Diverse: gli Eterodimeri
Sappiamo che gli mGluR possono formare eterodimeri, ad esempio mGluR2 può accoppiarsi con mGluR3, mGluR4 o mGluR7. Queste coppie diverse hanno sensibilità al glutammato e cinetiche di attivazione differenti. Ci siamo chiesti: come cambia il comportamento di mGluR2 a seconda del partner?
Usando sempre l’smFRET per monitorare la chiusura della conchiglia LBD di mGluR2, abbiamo scoperto che la velocità di apertura e chiusura cambiava drasticamente! Con mGluR3 o mGluR4 come partner, i movimenti erano più lenti e potevamo distinguere chiaramente gli stati aperto e chiuso. Con mGluR7 (o un altro mGluR2), i movimenti erano così rapidi da darci solo un segnale medio! Inoltre, la probabilità che la conchiglia di mGluR2 fosse chiusa dipendeva dal partner. È come se ogni partner “accordasse” finemente la dinamica dell’altro attraverso interazioni “orizzontali” all’interfaccia.
Ancora più sorprendente: abbiamo usato un agonista che attiva solo mGluR7 (LSP4-2022) nell’eterodimero mGluR2/7. Anche se mGluR2 non legava l’agonista, la sua conchiglia LBD tendeva a chiudersi più spesso! Questo dimostra che l’attivazione di una subunità può influenzare direttamente la conformazione del partner non legato.
Un Modello a Tappe per l’Attivazione
Mettendo insieme tutti questi pezzi – smFRET, MD, HDX-MS, studi su omo- ed eterodimeri – emerge un quadro complesso ma affascinante dell’attivazione degli mGluR. Non è un interruttore on/off, ma una serie di transizioni tra diversi stati conformazionali, con un accoppiamento lasco tra i movimenti dei vari domini (LBD, CRD, TMD).
Possiamo pensare a delle “porte” o “cancelli” lungo il percorso:
- Chiusura delle conchiglie LBD: Indotta dal glutammato, ma veloce e reversibile.
- Torsione degli LBD (cancello della torsione): Favorita quando entrambe le conchiglie sono chiuse, stabilizzata da interazioni specifiche (rete R177), ma ancora dinamica e non completa neanche a saturazione di glutammato. Qui si forma l’intermedio R-C/C.
- Avvicinamento dei CRD (cancello dell’attivazione): Segue la torsione LBD, ma con un ritardo. Popola stati progressivamente più vicini, favoriti da PAM e proteina G.
- Stato di accoppiamento alla proteina G: Lo stato CRD più vicino, raggiunto solo transitoriamente, che permette la segnalazione.
Questo modello a più stadi, con passaggi non perfettamente accoppiati, spiega la dinamicità del recettore e il suo ampio margine per la modulazione allosterica. Le differenze nelle interazioni all’interfaccia, sia verticali (tra domini) che orizzontali (tra subunità, specialmente negli eterodimeri), permettono alla famiglia mGluR di generare una gamma così vasta di risposte al glutammato, decodificando segnali su scale temporali e spaziali diverse.
Capire questi meccanismi nel dettaglio apre strade importantissime per disegnare nuovi farmaci che possano modulare finemente l’attività di specifici omo- o eterodimeri mGluR, offrendo potenziali terapie per disturbi neurologici e psichiatrici. Il ballo segreto di questi recettori è appena iniziato a svelarsi!
Fonte: Springer