Amiloidosi Ereditaria da Transtiretina (ATTRv-PN): Sveliamo i Segreti della Mutazione Ala97Ser in Cina
Ciao a tutti! Oggi voglio portarvi con me in un viaggio affascinante nel mondo della genetica e della neurologia, alla scoperta di una malattia rara ma insidiosa: l’Amiloidosi Ereditaria da Transtiretina con Polineuropatia (ATTRv-PN). In particolare, ci concentreremo su una specifica mutazione, la Ala97Ser (p.Ala117Ser), e sulle sue caratteristiche uniche osservate in una popolazione specifica della Cina meridionale continentale. Preparatevi, perché quello che abbiamo scoperto è davvero interessante!
Cos’è l’Amiloidosi Ereditaria da Transtiretina (ATTRv-PN)?
Prima di tuffarci nei dettagli della nostra ricerca, facciamo un passo indietro. L’ATTRv-PN è una malattia genetica rara, trasmessa con modalità autosomica dominante. Cosa significa? Che basta ereditare una copia del gene mutato da uno dei genitori per sviluppare la malattia. Il “colpevole” è il gene TTR, che si trova sul cromosoma 18. Questo gene produce una proteina chiamata transtiretina. Nelle persone con ATTRv-PN, a causa di una mutazione puntiforme (un piccolo errore nel codice genetico), questa proteina diventa instabile, si ripiega in modo anomalo e forma degli aggregati insolubili, chiamati fibrille amiloidi.
Queste fibrille si depositano in vari tessuti e organi, un po’ come una sorta di “ruggine” biologica, danneggiandoli progressivamente. I bersagli principali sono:
- Il sistema nervoso periferico (causando polineuropatia sensitivo-motoria)
- Il sistema nervoso autonomo (che controlla funzioni involontarie come battito cardiaco, digestione, pressione sanguigna)
- Il cuore
- I reni
- Gli occhi
Si tratta quindi di una malattia sistemica, che può manifestarsi con una vasta gamma di sintomi, spesso progressivi e invalidanti, che solitamente iniziano in età adulta. Pensate che la prima descrizione risale al 1952 in Portogallo! Da allora, è stata identificata in tutto il mondo, con oltre 130 diverse mutazioni del gene TTR conosciute.
La Mutazione Ala97Ser: Un “Marchio di Fabbrica” Cinese?
Ogni mutazione può dare origine a caratteristiche cliniche leggermente diverse. La mutazione più comune a livello globale è la Val30Met (p.Val50Met). Tuttavia, alcune mutazioni sono più frequenti in specifiche aree geografiche. È il caso della Ala97Ser (p.Ala117Ser), nota per essere comune tra le famiglie cinesi, specialmente nell’area di Taiwan.
La nostra ricerca si è concentrata proprio su questa mutazione, ma nella Cina meridionale continentale. Perché? Perché sembrava che questa variante fosse particolarmente diffusa lì, e volevamo capirne meglio le caratteristiche cliniche ed epidemiologiche. Abbiamo raccolto i dati di 21 pazienti provenienti da 20 famiglie diverse, diagnosticati con ATTRv-PN da mutazione Ala97Ser in tre grandi centri medici tra il 2013 e il 2024. Si tratta della casistica più ampia mai riportata finora per questa specifica mutazione in quest’area.
Una delle prime cose che abbiamo notato è un forte squilibrio di genere: 18 maschi e solo 3 femmine. Non siamo ancora sicuri del perché, potrebbe esserci un fattore protettivo nelle donne o un bias nella selezione dei casi, ma è un dato che salta all’occhio.

Il Quadro Clinico: Esordio Tardivo e Sintomi Predominanti
Un’altra caratteristica chiave emersa dal nostro studio è l’esordio tardivo della malattia. L’età media di comparsa dei primi sintomi nei nostri pazienti era di 56,5 anni (con un range tra 40 e 66 anni). Questo è diverso da altre mutazioni, come la Val30Met, che può manifestarsi sia precocemente che tardivamente.
Quali sono stati i primi campanelli d’allarme?
- Il sintomo iniziale più comune, riportato dal 71,4% dei pazienti (15 su 21), è stato il formicolio o l’intorpidimento (parestesia), tipicamente alle estremità (mani e piedi). Anzi, la parestesia era presente in tutti i pazienti al momento della diagnosi.
- Altri sintomi iniziali includevano debolezza alle gambe (2 pazienti), problemi gastrointestinali come diarrea/stitichezza (1 paziente), disfunzione erettile (1 paziente), ridotta tolleranza all’esercizio fisico (1 paziente) e senso di oppressione toracica (1 paziente).
Man mano che la malattia progrediva, il quadro si complicava. Abbiamo osservato:
- Debolezza muscolare e atrofia (perdita di massa muscolare): Presenti in 15 pazienti, soprattutto a livello distale (mani e piedi).
- Dolore agli arti: Riportato da quasi la metà dei pazienti.
- Riflessi tendinei diminuiti e sindrome del tunnel carpale: Molto comuni.
- Disfunzione del sistema nervoso autonomo: Riscontrata nell’85,7% dei pazienti (18 su 21). I sintomi variavano molto:
- Stipsi (11 pazienti, a volte alternata a diarrea).
- Perdita di peso significativa (17 pazienti).
- Ipotensione ortostatica (calo della pressione alzandosi in piedi, 7 pazienti).
- Disfunzione erettile (presente in 9 dei 18 pazienti maschi, spesso in fase precoce).
- Sudorazione eccessiva (iperidrosi, solo 4 pazienti).
- Problemi urinari (ritenzione, 2 pazienti).
- Coinvolgimento di altri organi:
- Cuore (80,9% – 17 pazienti): Il più frequente, con aritmie, ipertrofia cardiaca (ispessimento delle pareti del cuore) e insufficienza cardiaca sintomatica. Purtroppo, uno dei pazienti è deceduto per un evento cardiaco poco dopo la diagnosi.
- Reni (19,0% – 4 pazienti): Manifestato da alterazioni nella quantità di proteine nelle urine.
- Occhi (19,0% – 4 pazienti): Con sintomi come calo della vista, opacità vitreali (“mosche volanti”) e cataratta (ma nessun caso di glaucoma).
- Altri sintomi riportati includevano edema (gonfiore, 8 pazienti), tosse secca cronica (8 pazienti – un sintomo particolare già notato in un precedente caso con questa mutazione) e rash emorragico (macchie sulla pelle, 2 pazienti).
La Diagnosi: Un Percorso a Ostacoli
Gli esami strumentali hanno confermato il quadro. Gli studi di conduzione nervosa (NCS) hanno mostrato una polineuropatia sensitivo-motoria di tipo assonale (cioè un danno diretto alle fibre nervose, non alla loro guaina mielinica). Il danno era più severo agli arti inferiori e le fibre sensitive erano più compromesse di quelle motorie.
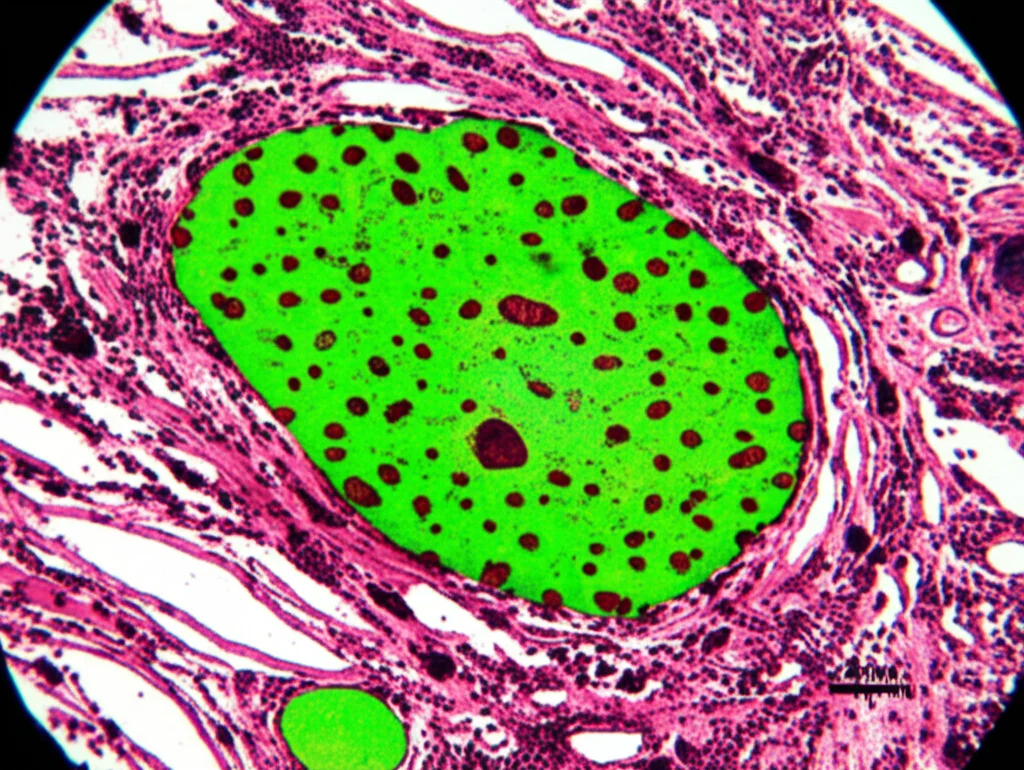
La biopsia del nervo (solitamente il nervo surale, a livello della caviglia) è stata eseguita su 15 pazienti. Nel 73,3% dei casi (11 su 15), la colorazione con Rosso Congo è risultata positiva, mostrando i tipici depositi di amiloide che appaiono di colore verde mela brillante se osservati al microscopio a luce polarizzata. Tuttavia, è importante notare che una biopsia negativa non esclude la malattia!
Nonostante questi strumenti, la diagnosi di ATTRv-PN rimane difficile. Pensate che ben 17 dei nostri 21 pazienti avevano ricevuto una diagnosi errata in precedenza! Gli errori più comuni?
- Polineuropatia Cronica Infiammatoria Demielinizzante (CIDP)
- Neuropatie periferiche di altra natura
- Ernia del disco cervicale o lombare
- Stenosi del canale spinale
Il ritardo diagnostico medio tra l’esordio dei sintomi e la diagnosi finale è stato di quasi 5 anni (4,9 ± 3,9 anni). Questo ritardo è un ostacolo enorme per una gestione ottimale della malattia, poiché esistono terapie (come il Tafamidis, che quasi tutti i nostri pazienti hanno iniziato, anche se non tutti hanno potuto proseguire per motivi di costo) che sono più efficaci se iniziate precocemente.
Un Effetto Fondatore e un Nuovo Modello di Valutazione
L’alta concentrazione di casi con mutazione Ala97Ser nella Cina meridionale continentale, insieme alle caratteristiche cliniche e demografiche osservate, ci fa ipotizzare un “effetto fondatore”. Significa che la mutazione potrebbe essere originata da un piccolo gruppo di antenati comuni in quest’area e poi trasmessa alle generazioni successive. Questo ha implicazioni importanti per capire la diffusione del gene e potrebbe facilitare lo screening dei portatori e la prevenzione.
Inoltre, abbiamo proposto un nuovo modello di punteggio multidimensionale, rappresentato graficamente con un “diagramma radar”. Questo strumento permette di visualizzare in modo più completo il coinvolgimento dei diversi sistemi (nervoso periferico, autonomo, cardiaco, renale, oculare) associato a specifiche mutazioni, andando oltre la classica dicotomia nervi/cuore. Speriamo che questo possa aiutare a caratterizzare meglio i diversi genotipi di ATTRv-PN e facilitare una diagnosi più precoce.

Cosa Portiamo a Casa?
Il nostro studio, pur con i limiti di uno studio retrospettivo e con un campione relativamente piccolo e geograficamente limitato, getta nuova luce sulle caratteristiche cliniche dell’ATTRv-PN associata alla mutazione TTR Ala97Ser nella Cina meridionale continentale. Abbiamo evidenziato:
- Un esordio tardivo.
- Una predominanza maschile.
- L’intorpidimento come sintomo iniziale più comune.
- Un elevato coinvolgimento del sistema nervoso autonomo e cardiaco.
- Un significativo ritardo diagnostico e un alto tasso di diagnosi errate.
- La possibile presenza di un effetto fondatore.
C’è ancora molta strada da fare per comprendere appieno questa malattia e migliorare la vita dei pazienti. Servono studi più ampi, multicentrici e prospettici. Ma ogni passo avanti nella conoscenza, come quello che abbiamo cercato di fare noi, è fondamentale per aumentare la consapevolezza, accelerare la diagnosi e offrire trattamenti tempestivi ed efficaci.
Spero che questo approfondimento vi sia stato utile e vi abbia incuriosito su questa complessa patologia!
Fonte: Springer





