A20: Lo Scudo Segreto dei Reni Contro l’Infiammazione da DNA ‘Danneggiato’
Ciao a tutti! Oggi voglio parlarvi di una scoperta affascinante che riguarda i nostri reni e come si difendono da un nemico insidioso che può nascere proprio dentro di noi. Parliamo di Insufficienza Renale Acuta (AKI), una condizione seria e potenzialmente letale in cui i reni smettono improvvisamente di funzionare a dovere.
Ma cosa scatena l’incendio? Il ruolo del DNA “ossidato”
L’AKI può avere molte cause, come infezioni gravi (sepsi) o l’uso di certi farmaci, ad esempio il cisplatino usato in chemioterapia. Indipendentemente dalla causa scatenante, spesso si innesca un circolo vizioso di morte cellulare, danno ai tessuti e, soprattutto, una fortissima infiammazione.
Qui entra in gioco un protagonista inaspettato: il nostro stesso DNA. Quando le cellule renali muoiono in modo incontrollato, rilasciano frammenti del loro DNA. Normalmente, il corpo ha sistemi per ripulire questi “detriti”, ma in condizioni di stress e infiammazione, questo DNA può subire un processo chiamato ossidazione. Immaginatelo un po’ come del metallo che arrugginisce: questo DNA autologo ossidato (ox-self-DNA) diventa più resistente alla degradazione e, peggio ancora, viene riconosciuto dal nostro sistema immunitario come un segnale di pericolo (un DAMP – Danger-Associated Molecular Pattern).
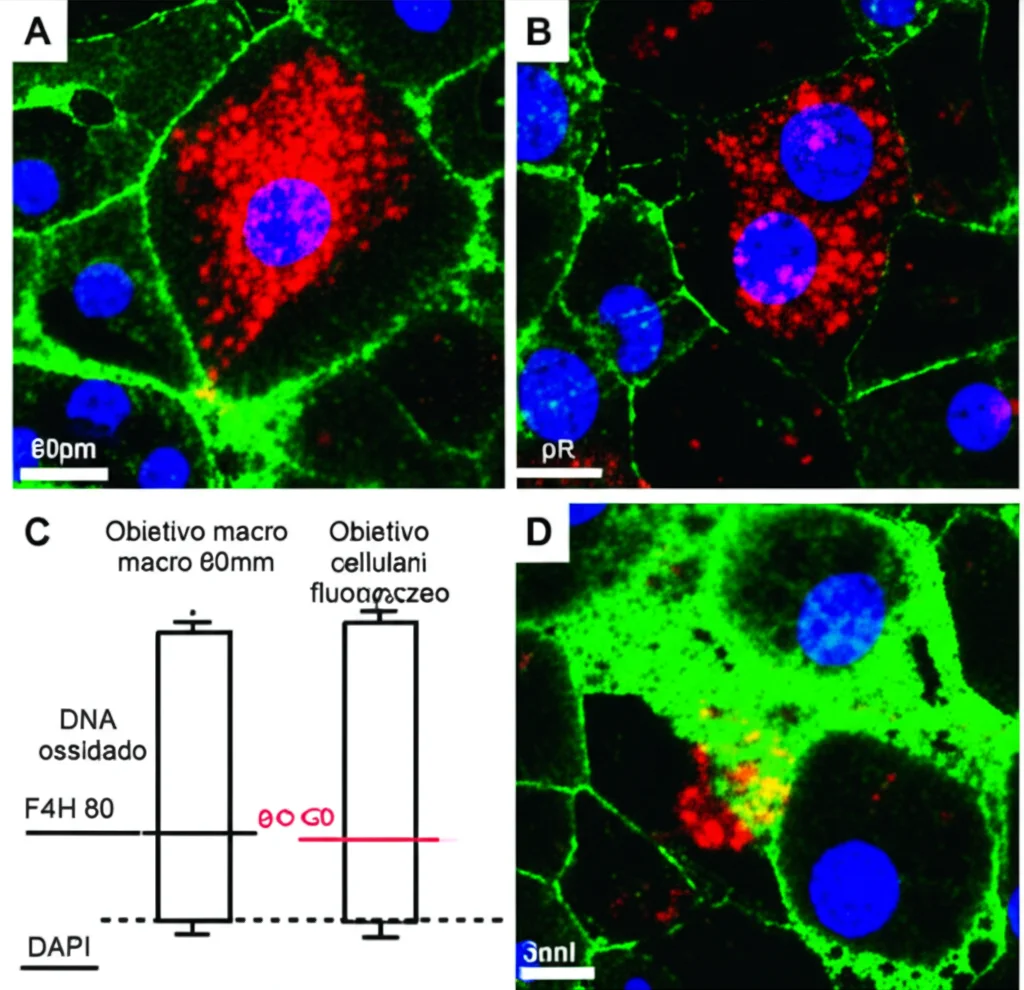
Abbiamo scoperto che questo DNA ossidato si accumula nel sangue sia dei pazienti umani con sepsi (una causa comune di AKI) sia nei topi in cui avevamo indotto l’AKI con diversi metodi (cisplatino, acido aristolochico I, lipopolisaccaride – LPS). Non solo, ma abbiamo visto che questo DNA “arrugginito” viene letteralmente “mangiato” dai macrofagi, le cellule spazzine del nostro sistema immunitario, proprio all’interno del tessuto renale danneggiato. Questo suggerisce che i macrofagi, una volta attivati da questo segnale, potrebbero amplificare ulteriormente l’infiammazione e il danno.
Due strade per l’infiammazione: STING e NLRP3
Ma come fa esattamente questo DNA ossidato a scatenare l’inferno infiammatorio? Abbiamo identificato due vie principali:
- La via cGAS-STING: Questo è un sistema di allarme cellulare che normalmente rileva il DNA virale. Quando attivato dal nostro DNA ossidato, produce interferoni di tipo I (IFN) e altre molecole pro-infiammatorie. Abbiamo visto che questa via si attiva nei topi con AKI e nelle cellule immunitarie (i macrofagi derivati dal midollo osseo, BMDM) trattate con il DNA ossidato estratto dai reni malati. Bloccare questa via (usando topi geneticamente modificati senza STING) ha portato a un leggero miglioramento della sopravvivenza e della funzione renale nei topi con AKI. Tuttavia, l’effetto era modesto.
- L’inflammasoma NLRP3 e la Piroptosi: Questa è la scoperta chiave! Il DNA ossidato ha una particolare affinità per attivare un complesso proteico chiamato inflammasoma NLRP3. L’attivazione di NLRP3 porta a una forma di morte cellulare programmata molto “infiammatoria” chiamata piroptosi (dal greco “pyro”, fuoco). Durante la piroptosi, la cellula si gonfia, si rompe e rilascia potentissime citochine infiammatorie come l’IL-1β e l’IL-18, oltre ad altri segnali di danno. Abbiamo osservato che il DNA ossidato induce potentemente la piroptosi nei macrofagi. E qui sta il punto cruciale: bloccare la piroptosi (usando un inibitore della proteina GSDMD, l’esecutore della piroptosi, chiamato disulfiram) o bloccare direttamente NLRP3 (con un farmaco chiamato MCC950) ha avuto un effetto molto più significativo nel migliorare la sopravvivenza e ridurre il danno renale nei topi con AKI, paragonabile all’effetto ottenuto rimuovendo il DNA con l’enzima DNase I.
Questo ci ha fatto capire che, sebbene la via STING contribuisca, è l’asse DNA ossidato -> NLRP3 -> Piroptosi il vero motore trainante dell’infiammazione devastante nell’AKI mediata da self-DNA.
A20: L’eroe che non ti aspetti
Di fronte a un’infiammazione così potente, il nostro corpo cerca di mettere in atto dei meccanismi di controllo, dei “freni” per evitare che la situazione degeneri completamente. Ci siamo chiesti: esiste una molecola che agisce come limitatore naturale in risposta al DNA ossidato?
Analizzando quali geni venivano attivati nei macrofagi esposti al DNA ossidato, abbiamo notato un aumento significativo dell’espressione di un gene chiamato Tnfaip3, che codifica per una proteina nota come A20. A20 è un enzima particolare, un “editor” dell’ubiquitina (una piccola molecola che etichetta le proteine per diverse funzioni), noto per il suo ruolo nel regolare l’infiammazione e mantenere l’equilibrio (omeostasi) cellulare.
Abbiamo scoperto che l’espressione di A20 aumenta proprio in risposta allo stimolo del DNA ossidato (e anche in risposta all’attivazione della via STING, suggerendo un collegamento tra le due vie infiammatorie). Questa produzione di A20 sembra dipendere principalmente dall’attivazione di un’altra via infiammatoria classica, quella di NF-κB.
Come A20 mette i bastoni tra le ruote all’infiammazione
Ma cosa fa esattamente A20 per calmare le acque? Abbiamo dimostrato che A20 agisce su entrambe le vie infiammatorie attivate dal DNA ossidato:
- Sulla via STING: Le cellule prive di A20 (nei topi geneticamente modificati, A20myel-KO, dove A20 è eliminato specificamente nelle cellule mieloidi come i macrofagi) mostravano una risposta STING esagerata al DNA ossidato. Al contrario, sovraesprimendo A20 nelle cellule, la via STING veniva smorzata.
- Sulla via NLRP3/Piroptosi: Questo è l’effetto più rilevante. Le cellule senza A20 erano molto più suscettibili alla piroptosi indotta dal DNA ossidato o da altri attivatori di NLRP3 (come LPS+ATP). Producevano più proteina GSDMD tagliata (la forma attiva che causa la piroptosi) e rilasciavano più IL-1β e segnali di danno cellulare (LDH). Al contrario, le cellule con più A20 erano più resistenti.
Questi effetti si sono confermati in vivo: i topi senza A20 nei macrofagi morivano prima e avevano danni renali peggiori quando veniva indotta l’AKI. Al contrario, aumentare i livelli di A20 nel rene (usando un virus adeno-associato, AAV) o stimolare la sua produzione (pre-trattando i topi con basse dosi di TNF-α, un noto induttore di A20) migliorava la sopravvivenza e riduceva il danno renale.
Il meccanismo nel dettaglio: A20, NEK7 e NLRP3
Come fa A20 a bloccare NLRP3 e la piroptosi in modo così efficace? La chiave sembra essere un’altra proteina chiamata NEK7. NEK7 è una chinasi (un enzima che aggiunge gruppi fosfato alle proteine) essenziale per l’attivazione di NLRP3. In pratica, NEK7 deve legarsi a NLRP3 perché l’inflammasoma si assembli correttamente e si attivi.
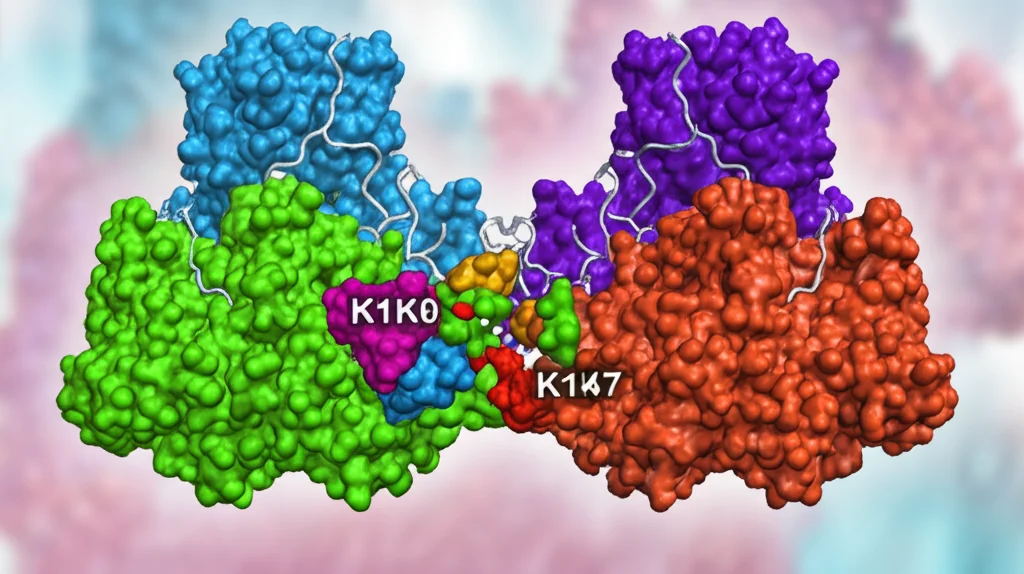
Studi precedenti (e anche i nostri dati di spettrometria di massa) avevano suggerito che A20 potesse legarsi a NEK7. Ora abbiamo approfondito questo legame. Usando tecniche di docking molecolare e generando mutazioni puntiformi su NEK7, abbiamo identificato un residuo specifico, la Lisina 140 (K140), come sito cruciale per il legame tra NEK7 e A20 (e anche per il legame con un piccolo peptide derivato da A20, chiamato P-II, che abbiamo sviluppato).
La cosa davvero interessante è che abbiamo scoperto che questo stesso sito K140 su NEK7 è anche fondamentale per il legame di NEK7 con NLRP3! Mutare K140 impediva a NEK7 di legarsi sia ad A20 sia a NLRP3. Questo suggerisce un meccanismo di competizione: A20 si lega a NEK7 nello stesso punto (o in un punto che interferisce) dove dovrebbe legarsi NLRP3. Legandosi a NEK7, A20 di fatto “sequestra” NEK7 o ne impedisce l’interazione con NLRP3, bloccando così l’attivazione dell’inflammasoma e la conseguente piroptosi.
Nuove speranze terapeutiche: mirare a NEK7
Questa scoperta apre scenari terapeutici molto promettenti. Se A20 protegge bloccando l’interazione NEK7-NLRP3, allora potremmo ottenere benefici terapeutici:
- Potenziando A20: Come abbiamo visto, aumentare A20 (con AAV o TNF-α) aiuta.
- Usando peptidi A20-mimetici: Il nostro peptide P-II, che mima la parte di A20 che lega NEK7, si è dimostrato efficace nel bloccare la piroptosi in vitro e, somministrato ai topi con AKI, ne ha migliorato significativamente la sopravvivenza e ridotto il danno renale.
- Bersagliando direttamente NEK7: Se NEK7 è il punto di snodo, cosa succede se lo eliminiamo o lo inibiamo?
- Abbiamo creato topi in cui NEK7 è eliminato specificamente nei macrofagi (Nek7myel-KO). Questi topi erano molto più resistenti all’AKI indotta da cisplatino: sopravvivevano più a lungo, avevano livelli di creatinina sierica (un marcatore di danno renale) più bassi e meno danni ai tubuli renali e fibrosi.
- Abbiamo anche “silenziato” NEK7 nei reni dei topi usando siRNA (piccoli RNA interferenti) recapitati con nanoparticelle, ottenendo risultati protettivi simili.
- Infine, abbiamo testato farmaci già noti per influenzare NEK7:
- La metformina (un comune farmaco per il diabete), che a basse dosi può ridurre l’espressione di NEK7, ha protetto i topi dall’AKI.
- La berberina (un composto naturale), nota per inibire l’attività di NEK7, ha anch’essa mostrato effetti protettivi significativi nei modelli di AKI.

Conclusioni: A20 e NEK7, guardiani del rene
In sintesi, il nostro lavoro svela un meccanismo affascinante e clinicamente rilevante: nell’Insufficienza Renale Acuta, il DNA rilasciato dalle cellule danneggiate e ossidato scatena una pericolosa infiammazione, principalmente attraverso l’attivazione dell’inflammasoma NLRP3 e la piroptosi. La proteina A20 emerge come un regolatore negativo cruciale, un vero e proprio scudo che limita questo processo infiammatorio agendo come un freno sulla via STING ma, soprattutto, interferendo competitivamente con il legame tra NEK7 e NLRP3.
Questa scoperta non solo ci aiuta a capire meglio cosa succede durante l’AKI, ma identifica anche nell’asse A20-NEK7-NLRP3 un bersaglio terapeutico estremamente promettente. Strategie mirate a potenziare A20, a usare peptidi A20-mimetici come P-II, o a inibire NEK7 (con siRNA o farmaci come metformina e berberina) potrebbero rappresentare nuove ed efficaci armi per combattere l’Insufficienza Renale Acuta e migliorare la prognosi dei pazienti. La ricerca continua, ma la strada sembra tracciata!
Fonte: Springer